
Although there exist a number of methods, such as NMR, X-ray, e.g., which explore the hydration of phospholipid bilayers, the solvent relaxation (SR) method has the advantage of simple instrumentation, easy data treatment and possibility of measuring fully hydrated samples. The main information gained from SR by the analysis of recorded “time-resolved emission spectra” (TRES) is micro-viscosity and micro-polarity of the dye microenvironment. Based on these parameters, one can draw conclusions about water structure in the bilayer. In this review, we focus on physical background of this method, on all the procedures that are needed in order to obtain relevant parameters, and on the requirements on the fluorescence dyes. Furthermore, a few recent applications (the effect of curvature, binding of antibacterial peptides and phase transition) illustrating the versatility of this method are mentioned. Moreover, limitations and potential problems are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
It was shown that the biological functions of many biomacromolecules and macromolecular bioassemblies depend on their hydration [1]. Among the others, the hydration of the phospholipid bilayers is still an open question. The water molecules in a membrane interact with functional groups of the amphiphilic phospholipids with various chemical natures resulting in a decently complex system. Several methods were employed to study the structure of hydrated phospholipid membranes and in particular NMR [2–4], fluorescence [5–7], X-ray and neutron diffraction [8,9] combined with theoretical simulations [10–12] provide the insight into the nature of the hydration. The results show that there is a gradient of the water concentration in the normal plane of the bilayer. The dependence is of a sigmoidal character featuring a steep decline at the glycerol region [13]. Water molecules form a clathrate hydration shell around the hydrophobic choline group [14]. On the other hand, they readily form H-bonds with the PO2 − group and with the C=O groups either within one lipid molecule or bridging neighboring lipid molecules [15,16]. These H-bonds appear to create an interfacial network providing another kind of attractive interactions stabilizing the bilayer. These H-bonds form and break with a frequency of 1011 s−1, and thus, the water molecules are attached to the phospholipid molecules almost instantaneously [17,18]. Surprisingly enough, several studies showed that the water molecules also penetrate down to the backbone region [19,20]. In addition, the increase in the water content in the headgroup region leads to the change in the alignment of the choline–phosphate dipole and in the lateral packing of the hydrocarbon region [21]. This finding gives a proof that the presence of water molecules effects the bilayer organization in a considerable way. Naturally, water molecules in the vicinity of the bilayer also show different properties compared to the bulk. It was reported that the strength, coordination number and lifetime of H-bonds is changed [22,23], the density is increased [24], and the motional freedom of water molecules is reduced in comparison to the bulk [25].
Among the fluorescence techniques employed, i.e., dynamical [26,27] and chemical [28] quenching [29], energy transfer [30,31], dye adsorption [32,33], lifetime (distributions) [34–36], and excimer formation [37,38], the determination of fluorescence anisotropy [34] has certainly been the dominating fluorescence method in studies of biological and model membranes. Fluorescence polarization studies, however, exhibit some major limitations. The membrane order parameters obtained from freely mobile probes like 1,6-diphenyl-1,3,5-hexatriene (DPH) result from a broad distribution of localization within the hydrophobic interior, the detailed characterization of which reveals inherent ambiguities [39]. Moreover, the anisotropy technique is limited to the characterization of the hydrophobic bilayer interior, since the anisotropy of headgroup labeled phospholipids appears to be rather insensitive to environmental changes [34]. These limitations do not restrict the application of the solvent relaxation method, the principles of which and some of their recent applications in membrane studies will be presented in this review. In this fluorescence method a very rapid electronic excitation of the fluorescence dye leads to the change in its charge distribution, however, according to Franck-Condon principle does not influence the position of the surrounding solvent molecules. Consequently, the solvent molecules are forced to adapt to the new situation and start to reorient in order to reach energetically favorable state. This dynamic process starting from the initial non-equilibrium “Franck-Condon state” and gradually proceeding toward the fully relaxed excited state is called solvent relaxation (SR). As the energy of the system is lowered, the relaxation process causes a continuous red-shift of the recorded time-resolved emission spectra. Thus, solvent polarity and the local environment have profound effects on the emission spectra of polar fluorophores. While interpretation of solvent-dependent emission spectra seems to be straight forward, a quantitative description of the solvent effects on fluorescence emission spectra is likely the most demanding topic in fluorescence spectroscopy. No single theory or type of interactions can be applied to all conditions. General solvent effects, which originate from the interactions of the dipole of the fluorophore with its environment, are coupled with specific fluorophore–solvent interactions, and often with complex photochemistry of the dye itself. Considering all these complexities one has to be very careful when processing and interpreting SR data. An additional caution is required when the SR experiment is performed in an isotropic and inhomogeneous media like biomembranes, where the SR occurs on the time–scale about four orders of magnitude longer than in the neat solvents.
Once properly applied SR method can provide valuable and unique information on the state of hydration of the system, and the influence of various factors on the hydration, such as peptide and protein binding [40, 41]. The history of fluorescence measurements of time-resolved emission spectra goes back almost 30 years ago [42], however, some crucial considerations related to SR processes in biomembranes have been omitted for a long time. This review presents an overview of a SR method applied to characterization of phospholipid membranes. In the following section, we will present the detailed description of the technique. Some important additional procedures and parameters, which can help to avoid some typical pitfalls, will be discussed (i.e., methods for inspection of extent of the observed relaxation process, namely time-zero estimation and examination of time-dependent width of emission spectra; choice of suitable fluorescent probes and their mean localizations; investigation of dye distribution; numerical characterization of the so called solvent response function). In the last section some of the recent applications of the method are presented.
METHODOLOGY
Time evolution of the emission of a suitable fluorescence probe carries complex data on its microenvironment. The most common method to extract this information is to analyze the time-resolved emission spectra (hereafter TRES). Following temporal changes of the spectrum position ν(t) (usually, ν is the frequency of the maximum of the TRES) and its width, the micropolarity and microviscosity of the dye environment can be characterized.
Parameters Yielded from TRES
Micropolarity/Hydration
The overall shift Δν, defined as:
is attributed to the difference between the energies of the Frank–Condon and the fully relaxed states, which are proportional to ν(0) and ν(∞), respectively [43,44]. It was shown, that Δν is directly proportional to the polarity function of a solvent, in which the dye is dissolved [44], since a more polar solvent leads typically to a stronger stabilization of the polar equilibrated excited state. The precise mathematical description of this relationship, however, depends on the choice of the dielectric solvation theory [45–52]. The fundamental “dielectric continuum solvation model” [50–52] predicts a linear proportionality between Δν and a dielectric measure of solvent polarity F for a large variety of solvents [44], where F is a function of the static dielectric constant (ɛ 0) and the optical refractive index (n) of the solvent.
Deviations from this linearity are found for a few aromatic solvents, indicating that F is a poor measure of the nuclear polarizability of aromatic solvents. However, for dielectric relaxation in phospholipid/water systems, it can be assumed that the Δν/F-proportionality is valid and that Δν value directly reflects polarity of the dye environment. In the biological and model membranes the polarity in the vicinity of the probe is determined mainly by the presence of the dipoles of water hydrating the membrane. Thus, Δν usually reflects the degree of hydration of the membrane at the level where the chromophore is located, giving the first major information provided by the solvent relaxation technique in the membrane studies.
Microviscosity/Mobility
The kinetics of the spectral shift depends on the mobility of the environment (i.e., reorientational motions of the solvent molecules), which in turn is determined by the frictional forces presented in the dye's surrounding. For quantitative evaluation of this kinetics it is convenient to normalize spectral response ν(t) to the overall shift as follows:
Correlation function C(t), called also spectral response function, is in the direct relation to the solvation dynamics of the solute (the dye). In fact, C(t) reflects the relaxation of naturally occurring fluctuations of the solvation energy δE from its average equilibrium value in the unperturbed system [53]:
The above relation was proved to be valid even for solutes, which undergo the protonation upon their electronic excitation, which illustrates the fact that the method probes the properties of the solvent, regardless of how severe the solute perturbation may be [53]. Although there have been several attempts to simplify the characterization of the SR process [54,55], the determination of the normalized spectral response function C(t) is certainly the most general and most precise way to characterize the time course of the solvent response. The easiest way to obtain numerical measure of the solvation kinetics is to integrate C(t) function over time:
Average relaxation time τ r, as defined in Eq. (5), was shown to be nearly proportional to the viscosity of the solvent [56–58]. It should be mentioned that this parameter, regardless of its convenience, gives only an approximate measure of the solvation kinetics and whenever more detailed description of the process is required, the correlation function itself should be analyzed. However one should choose correct model for fitting C(t). Commonly measured solvent relaxation processes, even in the neat solvents, are far from being monoexponential, while fitting C(t) for anisotropic media, such as phospholipids bilayers, often requires three and more exponential components. Multi-exponential fitting of experimental data needs theoretical assumptions about the model used, in order to be performed properly. It was shown that the residuals are often not enough to judge if the obtained time constants are meaningful [59]. Moreover, a user of the solvent relaxation technique should be aware that experimental factors like the signal-to-noise ratio, the excitation pulse width or the dynamics of the detectors influence the quality of the C(t) data. That is also why a fitting by physical models might appear as an over-interpretation of such data in some cases.
TRES Generation-Spectral Reconstruction Method
The C(t) function can be determined either by “single wavelength methods” [55,60] or, as most frequently, by “spectral reconstruction” [40,44,60–63], which requires a minimum of a priori assumptions and has been used for obtaining the results presented in this work. The primary data consist of a set of emission decays recorded at a series of wavelengths (typically 10–20) spanning the steady-state emission spectrum. These decays are fitted to a sum of exponentials using an iterative reconvolution technique until a mathematically satisfactory fit, is obtained [64]. It is important to note that the purpose of these fits is to obtain a parameterized form of the intensity decay D(t, λ) at each single wavelength, which is undistorted by the excitation pulse width and the time-dependence of the detecting electronics, and not to provide a physical interpretation of the decays. The TRES (S(λ, t)), at a given time t, are obtained by a relative normalization of those fitted decays D(t, λ) to the steady-state spectrum, S 0(λ):
The time dependence of the spectral shifts and the shape of the TRES are then used to determine the rates of relaxation and the nature of the relaxation process. The examination of the shape of the TRES can give information about the nature of the investigated relaxation process. The existence of isoemissive points [65], for example, indicates that the examined excited state process is not continuous but rather the existence of equilibrium between two excited states, like excimer or exciplex formation. In order to obtain desired parameters—time-dependent position of the spectrum ν(t) and its full width at half maximum (FWHM), the spectra should be transformed into wavenumber domain and fitted. Good choice for the fit function can be, for example, the log-normal line shape function [66], which gives a realistic picture of broad, asymmetric electronic emission bands [67, 68]:
where
The evolution of the fitted parameters (the peak height h, the peak position ν p, the asymmetry parameter, and the width parameter Δ), which are obtained via a nonlinear least-squares fitting method, fully describes the observed dynamics. Should the log-normal model fail to fit data, excited state processes other than pure continuous solvent relaxation may be considered.
Full Width at Half Maximum of TRES
The temporal behavior of the width of the time-resolved spectra provides rough but useful information on the extent of the observed solvent relaxation. The experiments carried out in phospholipid bilayers [61,70] and also in the supercooled liquids [69] have shown that the FWHM passes a maximum during solvation process. This is in a good agreement with the idea of a non-uniform spatial distribution of solvent response times [69,71]. It has been shown that in homogeneous systems of low molar mass molecules, the FWHM decays monotonically. In a spatially inhomogeneous systems however (i.e., where the microenviroments of the fluorophores differ), the relaxation behavior is different, since the solvent shells of individual fluorophores distributed in the system, respond with different rates to changes in the local electric field (which also spatially varies). This gives rise to a new phenomenon, which reflects the time distribution of phases of relaxations of individual solvation shells during the relaxation. The overall transient inhomogeneity increases significantly during the solvation and then decreases once the solvation finishes and the equilibrated excited state is reached. That is why the time-dependent width of TRES, which gives the measure of inhomogeneity of dyes microenvironments, passes a pronounced maximum. This effect gives the origin for the method of checking whether the entire response or merely part of it was captured within the time-window of the experiment. If only a decrease in the FWHM is observed, it means that the early part of the relaxation process is possibly beyond the time resolution of the equipment. In contrast, if only an increase is detected, the process is fairly slow under given conditions and the lifetime of the used fluorophore is not long enough to completely monitor the relaxation.
Besides, the above described application, the inspection of the time course of the FWHM is also an excellent tool to test whether a fluorescent probe has unimodal distribution of locations and how wide this distribution is. It is of particular importance in the view of recently published data concerning bimodal distribution of membrane dyes [72] as well as dye relocalization upon addition of ethanol [73] or change of the hydrostatic pressure [74]. The usefulness of this approach was illustrated by us during characterization of the solvent relaxation of the newly developed membrane label N-palmitoyl-3-aminobenzanthrone in different phase states of phospholipid bilayers [70]. All above gives the reason why FWHM profiles should always be examined in SR studies.
Time-Zero Estimation
To determine the correlation function C(t) from Equation (3) one needs to know the position ν(t = 0) of the so called “time-zero” spectrum—the hypothetical fluorescence emission spectrum of the molecules which are in the Frank–Condon state (i.e., are vibrationally relaxed but emit before any nuclear solvent motions have occurred). Such theoretical spectrum could not be directly observed even with ideal instrument with infinite time resolution [75]. One method to calculate this spectrum is the extrapolation of the measured spectra back to time t = 0. The extrapolated time-zero spectrum is unfortunately strongly affected by the experimental time resolution and its application often leads to significant errors in calculated solvation times [76]. As demonstrated for a series of membrane labels, a valid “time-zero” spectrum, is a pre-requisite for quantitative SR studies [70,73,77]. The solution for this problem was proposed by Fee and Maroncelli [76] and was based on the use of nonpolar reference spectra and a simple model of the inhomogeneous broadening of bands in polar solvents. The time-zero spectrum is estimated using only the emission and absorption steady-state spectra. The spectra measured in a non-polar reference solvent (such as hexane) are used to simulate spectral properties of the Frank–Condon state. According to the model of the electronic spectra of solvatochromic probes published in Reference [78], the method can be implemented as described below.
First, the absorption spectrum of the dye in the polar system of interest A p and absorption A ref(ν) and emission spectra F ref(ν) in the reference nonpolar solvent have to be measured. Then, the lineshape functions (g(ν)) and (f(ν)) for both reference spectra can be calculated:
The factors of ν in the denominators of Equations (9) and (10) are used so that f and g are directly proportional to the Einstein B coefficients for absorption and emission, respectively. Assuming that the spectra of individual molecules in different environments differ only by an overall frequency shift δ, the site distribution function can have the Gaussian profile:
where δ 0 is the average shift induced by the polar solvent and σ is the variance. The p(δ) can be calculated by fitting its convolution with g(ν) to the absorption spectrum A(ν), according to:
Once the distribution parameters are known, the desired time-zero spectrum F p can be computed from:
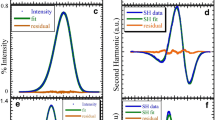
The ν ex in Equation (13) denotes the frequency of the monochromatic excitation. See Fig. 1. for the illustration of the method.

The estimated time-zero fluorescence emission spectrum of 4-[(n-dodecylthio)methyl]-7-(N,N-dimethylamino)-coumarin (DTMAC), (for structure see Fig. 2) (dotted line) and the spectra used for its calculation: Absorption g(ν) and emission f(ν) lineshape functions (solid lines) obtained from data measured in cyclohexane and absorption spectrum A p(ν) of the dye in DOPC LUVs (dashed line). All the spectra were measured in the room temperature. See text for details of the estimation procedure.
The absolute uncertainty of the above method was found to be less than 200 cm−1 [76]. In particular experiments, where the parameters values for similar systems are compared for which a part of time-zero estimation procedure is common, the relative uncertainties can be even smaller. The main drawback of the time-zero estimation is the need for reference spectra in a nonpolar solvent, which can be difficult to measure due to low solubility of some dyes. In this case measurements in frozen solvents, where the relaxation process is considerably slower, can serve as a good approximation. The estimation of the time-zero spectrum is essential for quantitative SR measurements. Knowing proper position of TRES at time t = 0 one can not only correctly calculate SR parameters (Equations (1) and (5)) but also calculate the percentage of the observed kinetics by comparison of the estimated ν(0) with the generated one. Thus, time-zero estimation tells us what part of the process occurs faster than the time resolution of our apparatus and is therefore missed. Only when there is strong evidence that the entire SR process takes place in the experimental time-window, the time-zero estimation procedure can be avoided.
When excitation is restricted to near the peak of the absorption and the underlying g(ν) is not too much structured, the estimation process can be significantly simplified:
where ν(abs) denotes wavelength for maximum of absorption and ν(em) for maximum of emission, subscript (p) stands for polar system of interest, and subscript (ref) for nonpolar reference. For excitation at the peak of the polar absorption spectrum the deviations between the frequencies determined using the complete method and those determined from Equation (14) are usually less than 50 cm−1 [76] giving total uncertainty for the simplified time-zero estimation equal 250 cm−1. Such accuracy will of course not be achieved for off-peak excitation or structured absorption lineshape. In fact even slightly structured spectrum, as shown on Fig. 1. leads already to the total uncertainty of 300 cm−1.
Membrane SR Probes and Their Location
As previously mentioned the dyes used in SR studies should be characterized by the large change in their dipole moment upon excitation (i.e., the charge distribution during excitation should change dramatically). Such dyes in addition to specific solvent–fluorophore interactions, can form an internal charge-transfer state (ICT) or a twisted internal charge-transfer (TICT) state [79] and often contain an electron donor and acceptor groups (for example amino and carbonyl groups respectively, but numerous other groups are known). Complex photochemistry and possible specific interaction between the probe and the molecules in its surrounding can significantly complicate the SR measurement.
Perhaps the most underestimated problem in the SR membrane studies, as well as in many other fluorescence techniques applied to the phospholipid bilayer, is the problem of the fluorophore location. It is of great importance to know where the fluorescence signal comes from. In other words, where, along the normal to the bilayer surface, is the probe located. In many studies the labeling is accomplished by simple partitioning of water-insoluble probes into the nonpolar regions of bilayer [80] leading to rather broad distribution of the dye in the backbone region or even worse, a coexistence of two probe populations in the membrane and in the bulk for lower partitioning coefficients. In that case, altering the properties of the membrane can simultaneously change the balance between dye populations. As both factors have influence on the SR behavior, they are usually impossible to separate. SR probes can be also covalently bound to the fatty acid chains or to the phospholipids. The depth of these probes in the bilayer can be adjusted by changing the placement of the fluorophore on the hydrocarbon chain as well as the length of the chain itself, resulting in more precise localization. The attachment of a trimethylammonium group, which is localized near the membrane–water interface, can give additional anchoring of the molecule and preserve its vertical orientation in the membrane. It is extremely important that the dye keeps its initial location during whole experiment or in all compared formulations. Even small change in the depth of the fluorophore will alter SR parameters drastically. This is because along the normal to the membrane a large polarity gradient exists plus SR kinetic changes from the value of hundreds of femtoseconds in the bulk to few nanoseconds at the glycerol level [63,77,81].
Figure 2 schematically presents a set of SR probes, which have been successfully used in our laboratory. The dyes locations are spanning the whole bilayer providing valuable tool for studying different membrane regions. Moreover, application of probes with identical but differently located fluorophores allows direct comparison between SR processes at different depths in the bilayer. Locations of some of the presented dyes were checked in quenching experiments with localized free radical quenchers according to the parallax method [82].

Distribution of used dyes, relative to the molecule of dioleoylphosphatidyl choline (DOPC), along the z-axis of phospholipid bilayer. From the left: DOPC, 11-((5-dimethylaminonaphthalene-1-sulfonyl)amino)undecanoic acid (Dauda) [77], 6,8-difluoro-4-heptadecyl-7-hydroxycoumarin (C17DiFU) [77], 4-[(n-dodecylthio)methyl]-7-(N,N-dimethylamino)-coumarin (DTMAC) [95], N-palmitoyl-3-aminobenzanthrone (ABA-C15) [70], 6-propionyl-2-dimethylaminonaphthalene (Prodan) [62,73,77,84], 6-lauroyl-2-dimethylaminonaphthalene (Laurdan) [77], 6-hexadecanoyl-2-(((2-(trimethylammonium)ethyl) methyl)amino)naphthalene chloride (Patman) [41,62,73,77,84], 2-(9-anthroyloxy)stearic acid (2-AS) [63,77], 9-(9-anthroyloxy)stearic acid (9-AS) [63,77], (16-(9-anthroyloxy)palmitoic acid (16-AP) [63].
TIME SCALE OF SR IN THE FLUID PHASE OF PHOSPHOLIPIDS BILAYERS [70,77,83]
Time-resolved spectroscopy is generally capable to monitor a particular process at a certain “time-window” that is covered by the experimental set-up. In case of monitoring TRES, ultimate time resolution of the instrument and the lifetime of the dye stand as the limiting factors. Herein, all reviewed experiments were recorded at the time-window of 50 ps to tens of nanoseconds. This (sub)-nanosecond time resolution is too slow for monitoring the solvation dynamics in the neat solvent. On the other hand, the SR process in viscous media or in the vicinity of supramolecular assemblies (i.e., micelles, vesicles, and proteins) is slowed down enough to be captured with the given (sub)-nanosecond “time-window”. Nevertheless, there is still high uncertainty if the whole process is captured with the available time resolution. Fortunately, the above described method of “time 0 estimation” providing the emission maximum of Franck–Condon spectrum serves as a quantitative tool to estimate how much of the SR process is missed. However, the relaxation process can also become substantially slower than the given time resolution. In this case, a time evolution of FWHM can serve as an instructive guide when considering the relative time scale of the experiment and the process of interest [81] as discussed in section “Full Width at Half Maximum of TRES”.
As far as the bilayers are concerned, the time scale of SR kinetics varies widely and is strongly dependent on the localization of a dye along the z-axis of the bilayer. It was demonstrated that bilayer might be separated into three regions that are characterized by different properties. Firstly, the dyes located in the external interface of the bilayer, which is formed by the bilayer surface and interacting water molecules from the bulk, show a complex behavior. For example, for dye 6,8-difluoro-4-heptadecyl-7-hydroxycoumarin embedded in the PC vesicles more than 50% of the solvent relaxation occurs on the shorter time scale than the given time resolution (50 ps) at 25°C [77]. The second part of the relaxation process, on the other hand, coincides with nanosecond “time-window”. Such an appearance might be explained by following considerations. A fluid phospholipid bilayer is a dynamic fluctuating system and broad statistical functions were found to be most suitable for describing the relative positions of the atoms along the z-axis of the bilayer. Similarly, the position of the dye molecules will also be spread over a significant region of the bilayer. Thus, at a certain time some part of the chromophore molecules might face an environment with a large amount of unbound free water which leads to the observed ultrafast solvation dynamics, whereas some ensemble of the dye molecules will be buried deeper probing nanosecond SR kinetics as discussed further. Secondly, a headgroup region, formed by the hydrated functional groups (i.e., choline, phosphate, glycerols and carbonyls), misses in the fluid phase the ultrafast (sub)-picosecond solvation dynamics almost completely and the SR kinetics occurs on the slower (sub)-nanosecond time scale [77,83]. It indicates that there is no free unbound water present in the vicinity of the dye. Moreover, the direct influence of the chemical composition on the solvation dynamics implies that the reorientational motions of the hydrated functional groups might be responsible for the substantial “slowing-down” of the solvation dynamics compared to the bulk. Fortunately, a set of dyes (Prodan, Laurdan, Patman) containing the identical chromophore enables to compare values of the dynamic Stokes shift, which is proportional to the micropolarity of the dye microenvironment. These dyes were found to be gradually located in the headgroup region which makes their use possible for mapping the solvation dynamics at various depths of the headgroup region [77]. It is evident that there is a polarity gradient in the headgroup region and the water content is increased moving from the headgroup region towards the bilayer interface [77,83]. Additionally, the SR kinetics becomes faster indicating higher mobility of the functional groups when approaching the interface [77,83]. Monitoring of SR behavior in the headgroup region by using a set of dyes Laurdan, Prodan and Patman was found to be most applicable. The reason is that substantial part (about 90%) of their SR response is resolved with the (sub)-nanosecond time resolution. Additionally, it seems that no additional (photophysical) process contributes to the time-dependent Stokes shift for the FWHM time evolution has not shown any anomaly up to now. Finally, the characterization of SR kinetics in the nonpolar, hydrophobic backbone region of fluid bilayers is complicated by the complex photophysics of the up to now only reachable dyes (“n-AS dyes”; for structures see Fig. 2). In addition to the SR process, a competitive intramolecular process (many hypothesis including the internal rotation, formation of TICT state and a role of the hydrogen bonding were proposed) makes the interpretation of TRES obtained for these n-AS dyes more complex and the conclusions have to be carefully drawn [77]. First of all, there is a steep decrease of the water concentration nearby the glycerol moiety, which is documented by a rather large difference in the Δν values obtained for 2-AS and 9-AS [77]. Secondly, water molecules are detected even close to the center of the bilayer (by 16-AP) [77], which is in contradiction to the results gained by the theoretical simulations [96]. This fact might be explained by the mechanism analogous to the preferred solvation observed at the interface of two immiscible liquids [97]. This mechanism proposes that the increase in the dipole moment caused by the excitation of the dye may draw the water molecules from the interface regions into the backbone region. By careful inspection of the TRES and considering the intramolecular relaxation process, we can draw the conclusion that the “intermolecular” SR kinetics becomes slower moving towards the bilayer center [77].
APPLICATIONS
The suitability of SR method for characterization of prothrombin binding to the phospholipid bilayer [85], its sensitivity to the chemical composition of lipid bilayer [81] and other applications [86,87] have already been reviewed several times [61,84]. Herein, we will introduce some recent applications of SR approach which will demonstrate its advantages and its potential drawbacks.
Influence of Temperature and Phase Transitions on SR [70]
The phase transition from the gel phase (L β ) to the liquid crystalline phase (L α ) is accompanied by the change of rigidity and by higher water penetration into the interior of the phospholipids bilayer. The SR method should naturally reflect these changes. Moreover, as reported earlier [40,62], the SR kinetics is supposed to differ significantly for both phases and in general, is supposed to turn faster with increasing temperature. The reason is that the motional freedom of the bilayer segments is increased as well. Having developed a new dye ABA-C15 (for chemical structure see Fig. 2) localized in the headgroup region, we decided to test these conclusions by monitoring solvation dynamics below, just above and far above the phase transition temperature T c [41].
The results obtained are summarized in Table I. show that the degree of hydration of the headgroup region is indeed being gradually increased when setting the experimental temperature further from the phase transition. Additionally, the mobility of the microenvironment was increased implying that the dye microenvironment became more mobile in the liquid crystalline phase, which is all in agreement with the above mentioned hypotheses. However, when investigating the SR data more thoroughly, additional conclusions might be assumed. First of all, FWHM time profile obtained below the phase transition for L β phase showed large deviation from the data observed for the fluid L α phase. There was an untypical decrease of FWHM at the initial times detected for L β phase (see Fig. 3) indicating that a second partially unresolved process was present. Moreover, despite of the fact that the overall integral time was slower, a smaller part of SR kinetics was captured for the rigid L β phase than for the “globally faster” L α phase. Such an anomalous behavior can be explained by a partial relocalization of the dye in the gel phase. When the bilayer is more packed, a number of the dye molecules are pushed away further from the headgroup region and consequently, this ensemble of dyes probes much faster solvation dynamics. This finding might elucidate why an ultrafast subpicosecond component of the solvation dynamics was reported below the phase transition temperature several times [88, 89]. It also shows the limitation of the SR method and one has to be aware that SR study covering more lipid phase states might suffer from the dye relocalization.

The time evolution of FWHM obtained for ABA-C15 at various phase states of the bilayer: (stars)—gel phase (DPPC); (circles)—liquid crystalline phase (close to the phase transition temperature) (DMPC); (triangles)—liquid crystalline phase (far above the phase transition temperature) (DOPC). Data were collected at 30°C.
Membrane Curvature [83]
In recent years an interest in studying membrane fusion has been raised especially in connection with the formation of tubules and vesicles inside cells [90], and with the formation of new organelles and enveloping of viruses [91]. The fusion is intermediated via highly deformed bilayer structures. For this reason we decided to reinvestigate the work based on a single wavelength approach [92] and to monitor the dependence of solvation dynamics in the headgroup region on the membrane curvature. Different membrane curvature of phospholipid bilayer was simulated by various vesicle dimensions. In case of LUVs (diameter 200 nm) the difference between the ratio of the inner and outer surface is approximately 1, whereas in case of SUVs (diameter 20 nm) it falls down to 0.5, which naturally influences the packing of the bilayer. In order to reveal these effects, we decided to label either both leaflets of the bilayer or the outer leaflet alone assuming that the selective labeling will affect the solvation dynamics in case of SUVs more dramatically than in case of LUVs. In order to get more detailed information on the curvature effect, three dyes; a coumarine like DTMAC, Prodan and Patman; which are gradually located towards the center of a bilayer (for their chemical structures and relative location within bilayer see Fig. 2), were used. Additionally, we also varied the lipid composition and performed the experiments for DOPC, DMPC, POPC and OPPC. The below described results, summarized for DOPC in Table II, appeared to be independent of the lipid composition. In conclusion, the higher curvature does lead to the different packing of the phospholipid bilayer, however, the effect is not as dramatic as estimated by a single-wavelength study using a dye with a rather undefined location [92]. It seems that the degree of hydration (Δν) is not modified by the higher curvature, nevertheless, the mobility of the dye microenvironment (τ r) is increased when the bilayer is more bent. This trend is clearly visible in the glycerol region (Patman) where the bound water is present exclusively. Approaching the interface region (DTMAC), which is accompanied by the gradual increase in a content of free water, these tendencies become less and less apparent. This study illustrates a strong point of the SR method enabling to characterize the properties of the dye microenvironment at various depths of the headgroup region.
Membrane Binding of Antibacterial Peptides [41]
In earlier studies it was reported that new insights into the nature of protein–membrane binding might be found by means of the SR method [40,85]. In a recent work we chose a headgroup dye, Patman, the location of which was reported to be invariant towards protein binding [40], and we decided to study the membrane-interaction of two antibacterial peptides, melittin and magainin, and one hydrophobic membrane-spanning sequence, KAL. We were interested in the impact of their binding on the dynamic properties near the glycerol moiety in the headgroup region [41]. All three peptides contain a positively charged and hydrophobic domain and their membrane binding is realized via hydrophobic and electrostatic interactions causing the permeabilization and/or disruption of the bacterial membrane [93].
The experiments were performed for neutral and negatively charged membrane containing biomimetic lipid, which was proved not to influence the SR behavior. The obtained data are concluded in Table III. It is demonstrated that the presence of the peptides modifies the solvation dynamics in the headgroup region. However, as far as magainin and melittin are concerned the changes in the micropolarity and viscosity at the glycerol region probed by Patman are significantly more pronounced when bilayer contains negatively charged lipids (POPC:DOPS mixture), as already observed in [94]. The SR kinetics is strongly slowed down which indicates that the mobility in the headgroup region is decreased significantly. Such an observation implies that the electrostatic interaction plays a role in peptide binding and causes a deeper insertion of the peptides into the bilayer. On the other hand, the changes detected for neutral lipids are smaller yet noticeable which draws the conclusion that the interaction is only of the interfacial nature. Another interesting fact is the observed difference between magainin and melittin binding. Whereas the interaction of magainin with a zwitterionic bilayer does not almost interfere the solvation dynamics in the headgroup region, the interaction of melittin leads to more significant changes in the SR. These findings imply, that hydrophobic interaction is more important in case of melittin, and it seems that the magainin is located at the external interface of the bilayer when bound to the neutral membrane. On the other hand, when a negative charge is introduced into a bilayer, magainin affects the headgroup region to a greater extent showing that the interaction is mainly maintained by the electrostatic forces. This behavior might also explain the reason for the selective targeting of magainin to the bacteria abundant in the negatively charged lipids. The hydrophobic sequence KAL shows the smallest effect of all the herein investigated peptides. It is probable that KAL inserts deepest to the hydrocarbon region [95] and that is why having almost no impact on the dynamics in the headgroup region. The presence of the negatively charged lipids also induces less apparent changes to the solvation dynamics indicating that the hydrophobic interaction is the driving force for KAL insertion. We believe that this peptide binding studies—together with the SR experiments on membrane binding of prothrombin [85]—demonstrate that the SR technique is very useful in mechanistic investigations of membrane–protein interactions.
CONCLUSIONS
When applying the SR method in bioassemblies, the exact knowledge of the location of the used dyes, the careful interpretation of the TRES, as well as the time-zero estimation are main prerequisites for valuable conclusions. On the other hand, the SR technique can give direct information on hydration and mobility in defined domains of fully hydrated bilayers. The validity of this approach can be demonstrated by a list of applications, like studies on lipid compositions [81,85] calcium ion concentrations [85], temperature [62,70,83], phase transitions [70,73], ethanol concentration [73], membrane curvature [83,92], as well as on the membrane binding of blood coagulation [85] and antibacterial proteins [41].
REFERENCES
J. A. Rupley and G. Careri (1991). Protein hydration and function. Adv. Protein Chem. 41, 37–172.
E. G. Finer and A. Darke (1974). Phospholipid hydration studied by deuteron magnetic-resonance spectroscopy. Chem. Phys. Lipids 12(1), 1–16.
T. Sparrman and P. O. Westlund (2003). An NMR line shape and relaxation analysis of heavy water powder spectra of the l-alpha, l-beta′, and P-beta′ phases in the DPPC/water system. Phys. Chem. Chem. Phys. 5(10), 2114–2121.
P. O. Westlund (2000). Line shape analysis of NMR powder spectra of (H2O)-H-2 in lipid bilayer systems. J. Phys. Chem. B 104(25), 6059–6064.
D. L. Bernik, D. Zubiri, E. Tymczyszyn, and E. A. Disalvo (2001). Polarity and packing at the carbonyl and phosphate regions of lipid bilayers. Langmuir 17(21), 6438–6442.
A. Chattopadhyay (2003). Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem. Phys. Lipids 122(1–2), 3–17.
S. Mazeres, V. Schram, J. F. Tocanne, and A. Lopez (1996). 7-Nitrobenz-2-oxa-1,3-diazole-4-yl-labeled phospholipids in lipid membranes: Differences in fluorescence behavior. Biophys. J. 71(1), 327–335.
S. Tristram-Nagle and J. F. Nagle (2004). Lipid bilayers: Thermodynamics, structure, fluctuations, and interactions. Chem. Phys. Lipids 127(1), 3–14.
M. C. Rheinstadter, C. Ollinger, G. Fragneto, and T. Salditt (2004). Collective dynamics in phospholipid bilayers investigated by inelastic neutron scattering: Exploring the dynamics of biological membranes with neutrons. Physica B Condens. Matter 350(1–3), 136–139.
P. Jedlovszky and M. Mezei (2001). Orientational order of the water molecules across a fully hydrated DMPC bilayer: A Monte Carlo simulation study. J. Phys. Chem. B 105(17), 3614–3623.
F. Zhou and K. Schulten (1995). Molecular-dynamics study of a membrane water interface. J. Phys. Chem. 99(7), 2194–2207.
I. Z. Zubrzycki, Y. Xu, M. Madrid, and P. Tang (2000). Molecular dynamics simulations of a fully hydrated dimyristoylphosphatidylcholine membrane in liquid-crystalline phase. J. Chem. Phys. 112(7), 3437–3441.
J. F. Nagle and S. Tristram-Nagle (2000). Structure of lipid bilayers. Biochim. Biophys. Acta Rev. Biomembr. 1469(3), 159–195.
M. Langner and K. Kubica (1999). The electrostatics of lipid surfaces. Chem. Phys. Lipids 101(1), 3–35.
A. Blume, W. Hubner, and G. Messner (1988). Fourier-transform infrared-spectroscopy of C-13=O-labeled phospholipids. Hydrogen-bonding to carbonyl groups. Biochemistry 27(21), 8239–8249.
J. Grdadolnik, J. Kidric, and D. Hadzi (1991). Hydration of phosphatidylcholine reverse micelles and multilayers—An infrared spectroscopic study. Chem. Phys. Lipids 59(1), 57–68.
H. Morgan, D. M. Taylor, and O. N. Oliveira (1988). Two-dimensional proton conduction at a membrane-surface—Influence of molecular packing and hydrogen-bonding. Chem. Phys. Lett. 150(3–4), 311–314.
M. Pasenkiewicz-Gierula, Y. Takaoka, H. Miyagawa, K. Kitamura, and A. Kusumi (1997). Hydrogen bonding of water to phosphatidylcholine in the membrane as studied by a molecular dynamics simulation: Location, geometry, and lipid–lipid bridging via hydrogen-bonded water. J. Phys. Chem. A 101(20), 3677–3691.
A. Chattopadhyay and S. Mukherjee (1999). Red edge excitation shift of a deeply embedded membrane probe: Implications in water penetration in the bilayer. J. Phys. Chem. B 103(38), 8180–8185.
E. Perochon, A. Lopez, and J. F. Tocanne (1992). Polarity of lipid bilayers—A fluorescence investigation. Biochemistry 31(33), 7672–7682.
V. Kurze, B. Steinbauer, T. Huber, and K. Beyer (2000). A H-2 NMR study of macroscopically aligned bilayer membranes containing interfacial hydroxyl residues. Biophys. J. 78(5), 2441–2451.
S. J. Marrink and H. J. C. Berendsen (1994). Simulation of water transport through a lipid-membrane. J. Phys. Chem. 98(15), 4155–4168.
S. J. Marrink, M. Berkowitz, and H. J. C. Berendsen (1993). Molecular-dynamics simulation of a membrane water interface—The ordering of water and its relation to the hydration force. Langmuir 9(11), 3122–3131.
J. F. Nagle and M. C. Wiener (1988). Structure of fully hydrated bilayer dispersions. Biochim. Biophys. Acta 942(1), 1–10.
U. Kaatze, A. Dittrich, K. D. Gopel, and R. Pottel (1984). Dielectric studies on water in solutions of purified lecithin vesicles. Chem. Phys. Lipids 35(3), 279–290.
A. P. Winiski, M. Eisenberg, M. Langner, and S. McLaughlin (1988). Fluorescence probes of electrostatic potential 1nm from membrane surface. Biochemistry 27(1), 386–392.
M. Langner, S. D. Cafiso, S. Marcelja, and S. McLaughlin (1990). The electrostatic properties of the phosphoinositides: Theoretical and experimental results. Biophys. J. 57(2), 335–349.
M. Langner and S. W. Hui (1993). Dithionite penetration through phospholipid bilayer as a measure of defects in lipid molecular packing. Chem. Phys. Lipids 65(1), 23–30.
M. R. Eftink (1992). in J. R. Lakowicz (Ed.), Topics in Fluorescence Spectroscopy: Principles. Plenum Press, New York, pp. 53–120.
B. W. Van der Meer, G. Coker, and S. Y. S. Chen (1994). Resonance Energy Transfer: Theory and Data. VCH Verlag, Weinheim.
R. Hutterer, F. W. Schneider, and M. Hof (1997). Anisotropy and lifetime profiles for n-anthroyloxy fatty acids: A fluorescence method for the detection of bilayer interdigitation. Chem. Phys. Lipids 86(1), 51–64.
M. Langner and S. W. Hui (1999). Merocyanine 540 as a fluorescence indicator for molecular packing stress at the onset of lamellar-hexagonal transition of phosphatidylethanolamine bilayers. Biochim. Biophys. Acta 1415(2), 323–330.
K. Kubica, M. Langner, and J. Gabrielska (2003). The dependence of fluorescein-PE fluorescence intensity on lipid bilayer state. Evaluating the interaction between the probe and lipid molecules. Cell Mol. Biol. Lett. 8(4), 943–954.
C. D. Stubbs and B. W. Williams (1992). in J. R. Lakowicz (Ed.), Topics in Fluorescence Spectroscopy: Biochemical Applications, Vol. 3. Plenum Press, New York, pp. 231–271.
C. Bernsdorff, A. Wolf, R. Winter, and E. Gratton (1997). Effect of hydrostatic pressure on water penetration and rotational dynamics in phospholipid–cholesterol bilayers. Biophys. J. 72(3), 1264–1277.
K. Brand, M. Hof, and F. W. Schneider (1991). Isotope effect in the time resolved fluorescence of anthracene in small unilamellar vesicles. Berichte Der Bunsen-Gesellschaft-Phys. Chem. Chem. Phys. 95(11), 1511–1514.
G. Duportail and P. Lianos (1996). in M. Rosoff (Ed.), Vesicles. Marcell Dekker, New York, pp. 296–372.
R. Hutterer, A. Haefner, F. W. Schneider, and M. Hof (1998). in J. Slavik (Ed.), Fluorescence Microscopy and Fluorescence Probes, Vol. 2. Plenum Press, New York, pp. 93–98.
U. A. van der Heide, G. van Ginkel, and Y. K. Levine (1996). DPH is localized in two distinct populations in lipid vesicles. Chem. Phys. Lett. 253(1–2), 118–122.
R. Hutterer, F. W. Schneider, W. T. Hermens, R. Wagenvoord, and M. Hof (1998). Binding of prothrombin and its fragment 1 to phospholipid membranes studied by the solvent relaxation technique. Biochim. Biophys. Acta 1414(1–2), 155–164.
T. Sheynis, J. Sykora, A. Benda, S. Kolusheva, M. Hof, and R. Jelinek (2003). Bilayer localization of membrane-active peptides studied in biomimetic vesicles by visible and fluorescence spectroscopies. Eur. J. Biochem. 270(22), 4478–4487.
J. H. Easter, R. P. Detoma, and L. Brand (1976). Nanosecond time-resolved emission-spectroscopy of a fluorescence probe adsorbed to l-alpha-egg lecithin vesicles. Biophys. J. 16(6), 571–583.
P. F. Barbara and W. Jarzeba (1990). in D. H. Volman, G. S. Hammond, and K. Gollnick (Eds.), Advances in Photochemistry, Vol. 15. Willey, New York, p. 1.
M. L. Horng, J. A. Gardecki, A. Papazyan, and M. Maroncelli (1995). Subpicosecond measurements of polar solvation dynamics—Coumarin-153 revisited. J. Phys. Chem. 99(48), 17311–17337.
B. Bagchi, D. W. Oxtoby, and G. R. Fleming (1984). Theory of the time development of the Stokes shift in polar media. Chem. Phys. 86(3), 257–267.
I. Rips, J. Klafter, and J. Jortner (1988). Solvation dynamics in polar liquids. J. Chem. Phys. 89(7), 4288–4299.
I. Rips, J. Klafter, and J. Jortner (1988). Dynamics of ionic solvation. J. Chem. Phys. 88(5), 3246–3252.
H. L. Friedman, F. O. Raineri, F. Hirata, and B. C. Perng (1995). Surrogate Hamiltonian description of solvation dynamics—Site number density and polarization charge-density formulations. J. Stat. Phys. 78(1–2), 239–266.
F. O. Raineri, B. C. Perng, and H. L. Friedman (1994). Surrogate Hamiltonian description of solvation dynamics—Resolution of global responses into spatial profiles. Chem. Phys. 183(2–3), 187–205.
N. G. Bakshiev (1964). Universal intermolecular interactions and their effect on the position of the electronic spectra of molecules in two-component solutions. Opt. Spectrosk. 16(5), 821–832.
Y. T. Mazurenko and N. G. Bakshiev (1970). Effect of orientation dipole relaxation on spectral and polarization characteristics of the luminescence of solutions. Opt. Spectosc. 28, 490–494.
W. Liptay (1974). in E. C. Lim (Ed.), Dipole Moments and Polarizabilities of Molecules in Excited States, Vol. 1. Academic Press, New York, pp. 129–229.
R. M. Stratt and M. Maroncelli (1996). Nonreactive dynamics in solution: The emerging molecular view of solvation dynamics and vibrational relaxation. J. Phys. Chem. 100(31), 12981–12996.
J. R. Lakowicz (1999). Principles of Fluorescence Spectroscopy, 2nd edn. Kluwer Academic/Plenum Press, New York.
I. Gonzalo and T. Montoro (1985). Interpretation of the fluorescence decay of 1-methylindole in polar-solvents by reorientational effects. J. Phys. Chem. 89(9), 1608–1612.
S. Arzhantsev, N. Ito, M. Heitz, and M. Maroncelli (2003). Solvation dynamics of coumarin 153 in several classes of ionic liquids: Cation dependence of the ultrafast component. Chem. Phys. Lett. 381(3–4), 278–286.
J. A. Ingram, R. S. Moog, N. Ito, R. Biswas, and M. Maroncelli (2003). Solute rotation and solvation dynamics in a room-temperature ionic liquid. J. Phys. Chem. B 107(24), 5926–5932.
N. Ito, S. Arzhantsev, M. Heitz, and M. Maroncelli (2004). Solvation dynamics and rotation of coumarin 153 in alkylphosphonium ionic liquids. J. Phys. Chem. B 108(18), 5771–5777.
A. A. Istratov and O. F. Vyvenko (1999). Exponential analysis in physical phenomena. Rev. Sci. Instrum. 70(2), 1233–1257.
V. Nagarajan, A. M. Brearley, T. J. Kang, and P. F. Barbara (1987). Time-resolved spectroscopic measurements on microscopic solvation dynamics. J. Chem. Phys. 86(6), 3183–3196.
M. Hof (1999). in W. Rettig, B. Strehmel, and S. Schrader (Eds.), Applied Fluorescence in Chemistry, Biology, and Medicine. Springer Verlag, Berlin, pp. 439–456.
R. Hutterer, F. W. Schneider, H. Sprinz, and M. Hof (1996). Binding and relaxation behaviour of Prodan and Patman in phospholipid vesicles: A fluorescence and H-1 NMR study. Biophys. Chem. 61(2–3), 151–160.
R. Hutterer, F. W. Schneider, H. Lanig, and M. Hof (1997). Solvent relaxation behaviour of n-anthroyloxy fatty acids in PC-vesicles and paraffin oil: A time-resolved emission spectra study. Biochim. Biophys. Acta Biomembr. 1323(2), 195–207.
M. Hof, J. Schleicher, and F. W. Schneider (1989). Time resolved fluorescence in doped aerogels and organosilicate glasses. Berichte Der Bunsen-Gesellschaft-Phys. Chem. Chem. Phys. 93(11), 1377
E. L. Wehry and L. B. Rogers (1966). in E. Hercules (Ed.), Fluorescence and Phosphorescence Analysis. Interscience, New York, pp. 81–149.
D. B. Siano and D. E. Metzler (1969). Band shapes of electronic spectra of complex molecules. J. Chem. Phys. 51(5), 1856–1859.
M. Maroncelli and G. R. Fleming (1987). Picosecond solvation dynamics of coumarin-153—The importance of molecular aspects of solvation. J. Chem. Phys. 86(11), 6221–6239.
E. W. Castner, M. Maroncelli, and G. R. Fleming (1987). Subpicosecond resolution studies of solvation dynamics in polar aprotic and alcohol solvents. J. Chem. Phys. 86(3), 1090–1097.
M. Yang and R. Richert (2001). Observation of heterogeneity in the nanosecond dynamics of a liquid. J. Chem. Phys. 115(6), 2676–2680.
J. Sýkora, V. Mudogo, R. Hutterer, M. Nepraš, J. Vaněrka, P. Kapusta, V. Fidler, and M. Hof (2002). ABA-C-15: A new dye for probing solvent relaxation in phospholipid bilayers. Langmuir 18(24), 9276–9282.
R. Richert (2001). Spectral diffusion in liquids with fluctuating solvent responses: Dynamical heterogeneity and rate exchange. J. Chem. Phys. 115(3), 1429–1434.
A. S. Klymchenko, G. Duportail, A. P. Demchenko, and Y. Mely (2004). Bimodal distribution and fluorescence response of environment-sensitive probes in lipid bilayers. Biophys. J. 86(5), 2929–2941.
R. Hutterer and M. Hof (2002). Probing ethanol-induced phospholipid phase transitions by the polarity sensitive fluorescence probes Prodan and Patman. Zeitschrift Fur Physikalische Chemie-Int. J. Res. Phys. Chem. Chem. Phys. 216, 333–346.
P. L. Chong, S. Capes, and P. T. T. Wong (1989). Effects of hydrostatic-pressure on the location of Prodan in lipid bilayers—A Ft-Ir study. Biochemistry 28(21), 8358–8363.
R. F. Loring, Y. J. Yan, and S. Mukamel (1987). Time-resolved fluorescence and hole-burning line-shapes of solvated molecules—Longitudinal dielectric-relaxation and vibrational dynamics. J. Chem. Phys. 87(10), 5840–5857.
R. S. Fee and M. Maroncelli (1994). Estimating the time-zero spectrum in time-resolved emission measurements of solvation dynamics. Chem. Phys. 183(2–3), 235–247.
J. Sýkora, P. Kapusta, V. Fidler, and M. Hof (2002). On what time scale does solvent relaxation in phospholipid bilayers happen? Langmuir 18(3), 571–574.
R. S. Fee, J. A. Milsom, and M. Maroncelli (1991). Inhomogeneous decay kinetics and apparent solvent relaxation at low-temperatures. J. Phys. Chem. 95(13), 5170–5181.
W. Rettig (1986). Charge separation in excited-states of decoupled systems—TICT compounds and implications regarding the development of new laser-dyes and the primary processes of vision and photosynthesis. Angewandte Chemie-Int. Ed. Eng. 25(11), 971–988.
D. Chakrabarty, P. Hazra, A. Chakraborty, and N. Sarkar (2003). Solvation dynamics of coumarin 480 in bile salt–cetyltrimethylammonium bromide (CTAB) and bile salt–Tween 80 mixed micelles. J. Phys. Chem. B 107(49), 13643–13648.
R. Hutterer, F. W. Schneider, and M. Hof (1997). Time-resolved emission spectra and anisotropy profiles for symmetric diacyl- and dietherphosphatidylcholines. J. Fluorescence 7, 27–33.
A. Chattopadhyay and E. London, (1987). Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26(1), 39–45.
J. Sýkora, P. Jurkiewicz, R. M. Epand, R. Kraayenhof, M. Langner, and M. Hof (2005). Influence of the curvature on the water structure in the headgroup region of phospholipid bilayer studied by the solvent relaxation technique. Chem. Phys. Lipids 135(2), 213–221.
R. Hutterer, A. Parusel, and M. Hof (1998). Solvent relaxation of Prodan and Patman: A useful tool for the determination of polarity and rigidity changes in membranes. J. Fluorescence 8(4), 389–393.
R. Hutterer and M. Hof (1996). Binding of prothrombin fragment 1 to phosphatidylserine containing vesicles: A solvent relaxation study. in J. Slavik (Ed.), Fluorescence Microscopy and Fluorescence Probes, Vol. 1. Plenum Press, New York, pp. 232–237.
M. Hof and P. Lianos (1997). Structural studies of thin AOT films by using the polarity fluorescent probe coumarin-153. Langmuir 13(2), 290–294.
H.-J. Egelhaaf, B. Lehr, M. Hof, A. Häfner, H. Fritz, F. W. Schneider, E. Bayer, and D. Oelkrug (2000). Solvation and solvent relaxation in swellable copolymers as studied by time-resolved fluorescence spectroscopy. J. Fluorescence 10(4), 383–392.
H. Bürsing, D. Ouw, S. Kundu, and P. Vöhringer (2001). Probing solvation dynamics in liquid water and at phospholipid/water interfaces with femtosecond photon-echo spectroscopies. Phys. Chem. Chem. Phys. 3(12), 2378–2387.
P. Dutta, P. Sen, S. Mukherjee, and K. Bhattacharyya (2003). Solvation dynamics in DMPC vesicle in the presence of a protein. Chem. Phys. Lett. 382(3–4), 426–433.
J. Zimmerberg and S. McLaughlin (2004). Membrane curvature: How BAR domains bend bilayers. Curr. Biol. 14(6), R250–R252.
W. B. Huttner and J. Zimmerberg (2001). Implications of lipid microdomains for membrane curvature, budding and fission—Commentary. Curr. Opin. Cell Biol. 13(4), 478–484.
M. Hof, R. Hutterer, N. Perez, H. Ruf, and F. W. Schneider (1994). Influence of vesicle curvature on fluorescence relaxation kinetics of fluorophores. Biophys. Chem. 52(2), 165–172.
K. Matsuzaki (1999). Why and how are peptide lipid interactions utilized for self-defense? Biochim. Biophys. Acta 1462(1–2), 1–10.
D. Allende, A. Vidal, S. A. Simon, and T. J. McIntosh (2003). Bilayer interfacial properties modulate the binding of amphipathic peptides. Chem. Phys. Lipids 122(1–2), 65–76.
J. A. Killian, I. Salemink, M. P. R. dePlanque, G. Lindblom, R. E. Koeppe, and D. V. Greathouse (1996). Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane alpha-helical peptides: Importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry 35(3), 1037–1045.
I. Z. Zubrzycki, Y. Xu, M. Madrid, and P. Tang (2000). Molecular dynamics simulations of a fully hydrated dimyristoylphosphatidylcholine membrane in liquid-crystaline phase. J. Chem. Phys. 112(7), 3437–3441.
D. Michael and I. Benjamin (2001). Molecular dynamics computer simulations of solvation dynamics at liquid/liquid interfaces. J. Chem. Phys. 114(6), 2817–2824.
ACKNOWLEDGMENTS
Financial support by the Czech Academy of Sciences (M.H., A.O., J.S, and P.J. via A400400503) and the Grant Agency of the Czech Republic (J.S. via 203/05/2308) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jurkiewicz, P., Sýkora, J., Ol żyńska, A. et al. Solvent Relaxation in Phospholipid Bilayers: Principles and Recent Applications. J Fluoresc 15, 883–894 (2005). https://doi.org/10.1007/s10895-005-0013-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-005-0013-4