
Abstract
Three cyclic polyoxothiomolybdates [Mo20O20S20(OH)20(PyD)2]4− (1) (PyD2− = 3,5-pyridinedicarboxylic acid, C7H6O4N2−), [Mo12O12S12(OH)12(H2O)5(NapH)]2− (2) (NapH2− = 1,4-naphthalene dicarboxylic acid, C12H6O42−) and [Mo8O8S8(OH)8(Ox)]2− (3) (Ox2− = oxalic acid, C2O42−) have been obtained from the condensation of the [Mo2O2S2]2+ building block in the presence of PyD2−, NapH2− and Ox2−, respectively. Compounds 1 − 3 were characterized by FT-IR spectroscopy, powder X-ray diffraction, UV–visible spectroscopy and thermogravimetric analysis. The structures of compounds 1 − 3 have been unambiguously established by single crystal X-ray crystallography. The electrochemical behaviors in DMF observed for three oxothiomolybdenum rings evidenced the ability to function as efficient electrocatalysts toward the reduction of protons into hydrogen.
Graphical Abstract
Three cyclic polyoxothiomolybdates self-assembled around carboxylate ligands were synthesized and characterized. The complexes showed electrocatalytic performance for HER in nonaqueous condition.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polyoxometalates (POMs) constitute a fascinating area of investigation, rich in thousands of inorganic compounds displaying various properties in catalysis, medicine [1, 2], magnetism [3, 4], conductivity [5, 6] or supramolecular chemistry [7, 8]. In recent years, due to their ability for electron storage, POMs play a significant role of efficient electro-catalysts for reduction of environmental pollutants or for energy conversion such as proton reduction into hydrogen [9,10,11].
Polyoxothiometalates (POTMs) belong to an under-studied subset of POM chemistry which emerged by the incorporation of chalcogens [12]. Oxygen bridges replaced by sulfur may alter their behaviour and chemistry which offers an opportunity for further exploration and findings. In particular, a few studies have shown that [Mo2O2S2]-based cyclic polyoxothiometalates (POTMs) are able to function as electro-catalysts toward the reduction of protons into hydrogen [13, 14]. This class of compounds are based on the self-condensation of [Mo2O2S2]2+ oxothiocations around anionic templates. Furthermore, this system was shown to be effective for developing extended cyclic inorganic clusters, differing in their nuclearity and shape by the nature of the encapsulated substrate, most commonly polycarboxylates [15,16,17,18,19,20,21,22]. However, the syntheses of such architectures requires theoretical support of driving and governing the formation of the host–guest assemblies [23,24,25]. In previous studies, linear saturated dicarboxylate ions gives highly flexible and fluxional host–guest architectures, while rigid templates like unsaturated or aromatic polycarboxylate ions are expected to bring rigidity to the host–guest system [15, 16, 18].
The electro-catalytic reduction of protons in DMF [14], in CH3CN [26, 27] and in aqueous medium [13] was observed for this class of molecular cycles. Earlier work of Keita et al. reported the ability of [Mo8Ox]2− (Ox2− = oxalate) and [Mo12Trim]3− (Trim3− = trimesic acid) to promote the reduction of protons into hydrogen by adding perchloric acid to DMF [14]. Hijazi and his coworkers have successfully designed and synthesized a new dodecamolybdenum wheel [Mo12O12S12(OH)12(Ni(dto)2)]2− (dto2− = dithiooxalato), the first example of a 3d transition metal embedded in the plane of the Mo ring, and the electrocatalytic properties for the reduction of protons was further confirmed in CH3CN [26]. The mechanism in the hydrogen evolution reaction (HER) process arisen from this class of molecular cycles has also been gradually understood in recent years, which may provide some optimization perspectives for the host–guest combinations. A proposed mechanism for HER catalyzed by [Mo12DMT]2−, [Mo12DFMT]2− and [Mo16DFMT]2− in aqueous medium highlighted the influence of the embedded ligand that the ligands incorporated in Mo-rings acts as a proton relay to some extent [28].
Herein, we report the syntheses and characterization of host–guest systems self-assembled around three carboxylate ions, namely, [Mo20-(PyD)2]4− (1), [Mo12-NapH]2− (2) and [Mo8-Ox]2− (3). Compound 1 is a helical icosanuclear ring constructed by 3,5-pyridinedicarboxylic acid as template, which is a monocyclic oxothiomolybdenum molecule with the highest nuclearity ever reported. Compound 2 is a new dodecanuclear pyriform ring constructed by 1,4-naphthalene dicarboxylic acid as template. Compound 3 is a well known structure and synthesized in a previously unreported way in this study. The resulting compounds were characterized by X-ray diffraction study and spectroscopic methods. Furthermore, the electrochemical behaviors were also investigated, indicating that three compounds are obviously active toward the reduction of protons.
Experimental Section
Reagents and Conditions
All reagents and chemicals were purchased from Energy Chemical or Nanjing Chemical Company Ltd. and used without further purification. (Me4N)2[Mo2O2S6] was synthesized according to the modified published procedure by E. Cadot et. al [29]. The [Mo2O2S2(H2O)6]2+ solution was prepared and stored under Ar and K2-x(Me4N)x[I2Mo10O10S10(OH)10(H2O)5]·20H2O (0 < x < 0.5) [30] was prepared as described in literature and characterized by routine methods.
Synthesis of [Mo20O20S20(OH)20(C7H6O4N)2]·72H2O (1)
K2-x(Me4N)x[I2Mo10O10S10(OH)10(H2O)5]·20H2O (0.50 g, 0.20 mmol) was suspended in 15 mL deionized water. 3,5-Pyridinedicarboxylic acid (43 mg, 0.25 mmol) was suspended in 10 mL deionized water and then slowly added to the former solution. The pH was adjusted to 5.0 using 1 M KOH after stirring for 20 min at room temperature. The resulting orange suspension was stirred for 30 min at 50 °C before filtration. Potassium chloride (0.75 g, 10 mmol) was then added to the clear filtrate with stirring for 20 min. After filtration, the clear orange solution was evaporated slowly in air at room temperature and provided yellow flake crystals after a week. (yield: 160 mg, 49% based on Mo). 1H NMR (400 MHz, DMSO-D6, ppm): δ 6.91 (s, 4H), 5.86 (s, 2H). IR (KBr disc): ν (cm) = 1614 (s), 1558 (s), 1481 (ms), 1380 (s), 1288 (w), 1122 (ms), 948 (s), 732 (ms), 524 (s). Anal. calc. for −1C14H150N2O120S20Mo20: C, 3.48; H, 3.13; N, 0.58; S, 13.28. Found: C, 3.35; H, 3.47; N, 0.65; S, 13.11%. TGA: 9% weight loss between 25 ~ 120 °C, corresponding to the removal of residual water molecules.
Synthesis of K1.13Cs0.87[Mo12O12S12(OH)12(H2O)6(C12H6O4)]·12H2O (2)
5 mL [Mo2O2S2(H2O)6]2+ (0.14 mol/L) solution was diluted to 8 mL by deionized water. 1,4-naphthalene dicarboxylic acid (60 mg, 0.25 mmol) was dissolved into 10 mL deionized water and KOH (28 mg, 0.50 mmol) was then added before stirring for 5 min. The resulting solution was slowly added to the diluted red solution dropwise and stirred for additional 20 min at room temperature, with a few yellow solid precipitated. The pH was adjusted to 5.0 using 1 M KOH and the solids increased gradually. The orange suspension was stirred for 30 min at 50 ℃ before filtration. Cesium chloride (0.75 g, 10 mmol) was then added to the clear filtrate with stirring for 20 min. After filtration, the clear orange solution was evaporated slowly in air at room temperature and provided yellow flake crystals after a week. (yield: 40 mg, 30% based on Mo). 1H NMR (400 MHz, DMSO-D6, ppm): δ 8.77 (s, 2H), 7.74 (s, 2H), 7.46 (s, 2H). IR (KBr disc): ν (cm−1) = 1637 (ms), 1577 (ms), 1392 (ms), 1313 (ms), 1079 (w), 948 (s), 786 (ms), 514 (s). Anal. calc. for C12H32O46S12Cs0.87K1.13Mo12: C, 5.53; H, 1.01; S, 14.77. Found: C, 5.87; H, 1.35; S, 15.08%. TGA: 12.2% weight loss between 25 ~ 150 °C, in agreement with the removal of 17 water molecules (calcd 12.5%).
Synthesis of K(NMe4)[Mo8O8S8(OH)8(C2O4)]·6H2O (3)
K2-x(Me4N)x[I2Mo10O10S10(OH)10(H2O)5]·20H2O (0.50 g, 0.20 mmol) was suspended in 15 mL deionized water. Oxalic acid (43 mg, 0.25 mmol) was dissolved in 10 mL deionized water and then slowly added to the former solution dropwise with stirring for 20 min at room temperature. The pH was adjusted to 4.5 using 1 M HCl after stirring for 20 min at room temperature. The resulting orange suspension was stirred for 30 min at 50 °C before filtration. Potassium chloride (0.75 g, 10 mmol) was then added to the clear filtrate with stirring for 20 min. After filtration, the clear orange solution was evaporated slowly in air at room temperature and provided yellow flake crystals after a week. (yield: 120 mg, 20.5% based on Mo). IR (KBr disc): ν (cm−1) = 1640 (s), 1540 (s), 1430 (ms), 1370 (ms), 1110 (ms), 947 (s), 849 (w), 750 (w), 519 (s). Anal. calc. for C6H32O28NS8KMo8: C, 4.42; H, 1.98; N, 0.86; S, 15.71. Found: C, 4.38; H, 2.06; N, 0.86; S, 15.83%. TGA: 6.7% weight loss between 25 ~ 130 °C, in agreement with the removal of 8 water molecules (calcd 6.6%).
Results and Discussion
Syntheses and Structures
The syntheses of compounds 1 − 3 correspond to now well-established procedures (Scheme 1). The condensation reaction of [Mo20-(PyD)2]4− (1) and [Mo8-Ox]2− (3) were performed by addition of potassium hydroxide until pH = 5.0 or hydrochloric acid until pH = 4.5 to an aqueous suspension containing the precursor K2−x(NMe4)x[I2Mo10O10S10(OH)10(H2O)5]·20H2O (0 < x < 0.5) and 3,5-pyridinedicarboxylic acid or oxalic acid, respectively. The synthesis of the anionic cluster [Mo12-NapH]2− (2) was performed by addition of potassium hydroxide until pH = 5.0 to an aqueous solution containing the thiometalate dimer [Mo2O2S2(H2O)6]2− solution and K2NapH. The potassium salts K2NapH was priorly prepared by deprotonation of 1,4-naphthalenedicarboxylic acid by a stoichiometric amount of potassium hydroxide in water. However, the conditions of precipitation are determinant for the isolation of these oxothiomolybdenum wheels. Precipitation by using potassium chloride yields [Mo20-(PyD)2]4− (1) and [Mo8-Ox]2− (3) as yellow crystals, and [Mo12-NapH]2− (2) can be crystallized as a cesium salt to give single crystals suitable for X-ray diffraction study. Powder X-ray diffraction patterns reveal that the positions of the characteristic peaks basically correspond to simulated spectrum, verifying the purity of compounds (Fig. S2).

Synthetic routes of oxothiomolybdenum rings 1−3
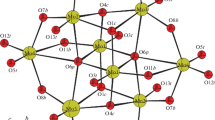
The molecular structures of compounds 1 and 3 are depicted in Figs. 1 and 2, respectively, and their crystal data are given in Table 1. The solid-state structure of compound 2 was also determined and was found to be with the formulation of K1.13Cs0.87[Mo12O12S12(OH)12(H2O)6(C12H6O4)]·12H2O though its crystal quality is bad due to disorder of aquo ligands in the inner cavity. Compound 1 crystallizes in the C2 space group, and the [Mo20(PyD)2]4− polythioanion structure is a helical icosanuclear ring encapsulating two PyD2− anions. It is known to be a monocyclic molecule with the highest nuclearity ever reported. The ten [Mo2S2O2]2+ building blocks are connected through double hydroxo bridging ligands, which span nonbonding Mo∙∙∙Mo contacts [3.344(2) ~ 3.358(2) Å] alternating with short Mo − Mo bonding contacts within the building blocks [2.845(2) ~ 2.851(2) Å] as usually observed for oxothiomolybdenum rings [31]. The molybdenum atoms of the ring are each bound to a terminal oxo group [1.638(16) ~ 1.740(15) Å] and are alternatively bridged by two hydroxo ligands [1.997(16) ~ 2.167(15) Å], two sulfide bridges [2.268(6) ~ 2.351(6) Å]. The two PyD2− moieties are asymmetrically anchored to the inorganic host with Mo − O distances in the 2.28(3) ~ 2.33(2) Å range, each oxygen atom bridging one adjacent Mo atom. Besides, each nitrogen atom in pyridine is singly bound to Mo atom [Mo5 − N1 = 2.31(3) Å], which agrees with the [Mo2O2S2(EDTA)]2− anion reported in the literature [32, 33]. Interestingly, it may be inferred to account for anti-conformation and certain distortion of the ring. The Mo − N bonds remove the plane structure to form a spontaneous chiral structure, where the individual molecular is chiral. It may be an effective strategy to construct similar high-nuclear cyclic molecules using pyridine ligands as templates. Besides, there are no coordinated water molecules in the structure due to the steric hindrance effect and hydrophobic effect of ligands. Consequently, ten Mo atoms connected with the ligands display an octahedral environment, while the other ten Mo atoms exhibit a square pyramidal geometry, the connections between each building unit are edge sharing (Fig. 1c).

Top (a) and side (b) views of labelled ball-and-stick representations of 1. Polyhedron mode of 1 (c). Space-filling representation of 1 (d). Color code: Mo in blue, oxygen in red, sulfur in yellow, carbon in grey and nitrogen in dark blue (Color figure online)

Top (a) and side (b) views of labelled ball-and-stick representations of 3. Polyhedron mode of 3 (c). Space-filling representation of 3 (d). Color code: Mo in blue, oxygen in red, sulfur in yellow and carbon in grey (Color figure online)
The introduction of the 1,4-dicarboxylnaphthalene as a template for the [Mo2S2O2]2+ self-condensation forms dodecanuclear pyriform planar ring [Mo12-NapH]2− (2) (Fig. S1). The bond lengths of molybdenum atoms to terminal oxo groups [1.60(3) ~ 2.33(4) Å], hydroxo ligands [1.97(3) ~ 2.22(3) Å] and sulfide bridges [2.233(14) ~ 2.319(14) Å] are similar to those of compound 1. Conversely to compound 1, compound 2 contains only one ligand conventionally anchored to the inorganic moiety through the carboxylate groups and two carboxylate groups are bound to Mo6 and Mo11 through O1 and O6, respectively [Mo6 − O11 = 2.30(3) Å; Mo11 − O25 = 2.28(3) Å], and bridged to Mo4 and Mo5 or Mo1 and Mo12 through O2 or O5, respectively [Mo4 − O10 = 2.26(2) Å; Mo5 − O10 = 2.39(3) Å; Mo1 − O26 = 2.39(3) Å; Mo12 − O26 = 2.20(3) Å]. As observed for the NapH2− ligand, steric hindrance imposes the aromatic ring of NapH2− ligand to be nearly perpendicular to the plane of metal rings. The inner cavity also contains five aquo ligands.
The oxothiomolybdenum anion skeleton is similar to that previously reported [16].The oxalic acid was introduced to the [Mo2S2O2]2+ self-condensation in the formation of compound 3 which crystallises in the P21/c space group (Fig. 2). The inorganic skeleton of the octameric ring (Fig. 2a) results from the self-linking of four [Mo2S2O2]2+ building blocks. Each Mo center is octahedral, which is bound to a terminal oxo group [Mo = Ot = 1.671(3) ~ 1.691(3) Å] and bridged by a oxo group of carboxyl ligand [2.475(3) ~ 2.518(3) Å], two hydroxo ligands [2.063(3) ~ 2.094(3) Å], and two sulfide bridges [2.3065(10) ~ 2.3236(10) Å]. The Ox2− anion is in central cavity, bonded to eight adjacent Mo atoms through Mo − μ2 − O interactions. The compound can be described as a ring of alternately edge and face-sharing octahedra (Fig. 2c). A S–S edge links the two octahedra of the same building block, and the building blocks are linked through common faces consisting of two OH ligands and one oxygen on a carboxyl group.
Infrared Spectra
FT-IR spectra of compounds 1 − 3 are depicted in Fig. S3. Each spectrum exhibits a broad band at around 3400 cm−1 associated with the water molecules. The strong absorptions in the spectrum of compound 1 observed at 1122 cm−1 ~ 1614 cm−1 correspond to stretching vibrations of C = C, C = N, C = O and C − H in two PyD2− ligands. Similarly, the absorptions in the spectra of compound 2 observed at 1079 cm−1 ~ 1637 cm−1 correspond to stretching vibrations of C = C, C = O and C − H in NapH2− ligand, and the absorptions in the spectra of 3 observed at 1100 cm−1 ~ 1640 cm−1 correspond to stretching vibrations of C − C, C = O, C − O and C − H in Ox2− ligand. The features of three compounds at 930 ~ 970 cm−1, 700 ~ 800 cm−1, 500 ~ 550 cm−1 are assigned to stretching vibrations of Mo = O, Mo − O − Mo and Mo − S − Mo in the inorganic oxothiomolybdenum rings.
Thermal Analysis
The TGA curves for three compounds were recorded in the presence of N2 gas at a heating rate of 5 °C min−1 and show three significant weight losses in the 25 ~ 800 °C temperature range (Fig. S4). The first stage corresponds to the loss of adsorbent water molecules and lattice water molecules, in which compound 1 has a total weight loss of 9% at 25 °C ~ 120 °C and compound 2 has a total weight loss of 10.6% at 25 ~ 130 °C and compound 3 has a total weight loss of 6.7% at 25 ~ 130 °C. The second stage is attributed to the loss of the external cations and carboxylate ligands in the inner cavities, in which compound 1 has a total weight loss of 17.8% at 333 °C ~ 525 °C and compound 2 has a total weight loss of 10.2% at 228 °C ~ 414 °C and compound 3 has a total weight loss of 6.76% at 175 °C ~ 417 °C. The prior decomposition of the ligands and the external cations, of which main products are CxHy, NH3, and CO2 [33], confirms the relative stability of Mo-ring moieties. The third weight loss is ascribed to the collapse of the oxothiomolybdenum rings with a partial desulfurization. As the temperature was raised, a gradual decrease of weight loss of compound 1 is observed from 766 °C to 800 °C without ending, whereas the decomposition of compound 2 or 3 is observed at 417 °C ~ 759 °C or 468 °C ~ 764 °C with a total weight loss of 11.9% or 9%.
Stability Studies
Prior to electrochemical studies, the stability of compounds 1 − 3 were studied in DMF by UV–visible spectroscopy (Fig. S5). In these cases, the obtained spectra remained identical after 24 h, suggesting that 1–3 are perfectly stable under such conditions. These results are further supported by 1H NMR. The 1H NMR spectra of compounds 1 and 2 in DMSO at room temperature are depicted in Fig. S6. For compound 1, two singlets at 5.86 and 6.91 ppm, with integration of 2 and 4 respectively, are assigned to two PyD2− encapsulated within the Mo20-ring. The NapH2− encapsulated within the Mo12-ring is observed as three signals at 7.46, 7.74 and 8.77 ppm with integration 2:2:2.
The stability of 1–3 in DMF solutions containing 0.1 M Bu4NPF6 were checked by UV–visible spectroscopy upon stepwise additions of trifluoroacetic acid (TFA). As shown in Fig. S7, similar absorptions for the three compounds come at 280 and 350 nm, corresponding to E → Mo (E = O, S) ligand-to-metal charge transfer [14, 34]. For the three compounds, the stability in solution in the presence of acid was evidenced with the increasing amount of acid, expressed as γ (γ = [acid]/[POTM]).
Electrochemical Studies
The cyclic voltammograms (CVs) of compounds 1 − 3, recorded at a static glassy carbon working electrode in DMF solutions containing 0.1 M Bu4NPF6 as electrolyte, are shown in Fig. 3. X-ray photoelectron spectroscopy (XPS, Fig. S8) measurements indicated that the Mo 3d spectra of three compounds all present Mo5+ 3d3/2 and Mo5+ 3d5/2 peaks at about 230 and 240 eV, which was conducive to further knowledge on the redox process. The cyclic voltammogram of compound 1 displays two defined irreversible waves, a monoelectronic reductive irreversible event around Epc = − 0.843 V vs Ag/AgCl attributed to the reduction of some Mo(V) centers of the ring and a multielectronic irreversible oxidation process located at Epa = + 0.811 V vs Ag/AgCl attributed to the oxidation of some bridging sulfur of the ring, in agreement with the previously related reports [13, 35]. The cyclic voltammogram of compound 2 exhibits the same pattern. The reductive irreversible process of 2 is centered around Epc = − 0.862 V vs Ag/AgCl arising from the reduction of Mo(V) centers and the oxidation irreversible process associated with the oxidation of bridging sulfur is located at Epa = + 0.804 V vs Ag/AgCl. Compared with compounds 1 and 2, the CV of compound 3 exhibits the similar behaviour except for another irreversible oxidation process located at more positive potentials, containing a contribution arising from the oxidation of Mo(V) centers. Remarkably, the scan rate did not affect the intrinsic electrochemical behavior of 1 − 3, but the potentials of anodic peak or cathodic peak were slightly shifted to more positive or negative values, respectively, as the scan rate increased. The current intensity of anodic peaks or cathodic peaks varies linearly with the scan rate, suggesting the reversible electron transfer is surface-confined [36].

Cyclic voltammograms of the compounds 1 (a), 2 (b) and 3 (c) in DMF + 0.1 M Bu4NPF6. Inset: linear dependence of the catalytic current with the scan rates (Scan rate from 50 mV s−1 to 400 mV s−1). Comparison of the cyclic voltammograms for the blank and compounds 1 − 3 (d) (Scan rate was 100 mV s−1)
Electrocatalytic Properties
Figure 4 shows the cyclic voltammograms of compounds 1 − 3 in DMF + 0.1 M Bu4NPF6, upon stepwise additions of TFA (pKa = 12.7 in DMF). The addition of increasing amounts of TFA in a solution of compound 1 leads to the growing of a new composite cathodic wave at about Epc = − 1.118 V vs Ag/AgCl and grows gradually with the excess parameter γ (below 10). This wave is well-separated from the formerly observed waves, and such a reduction process is attributed to the catalytic reduction of protons. Regardless of the potential scan rate, this wave remains chemically irreversible and does not affect the intrinsic electrochemical behavior of compound 1. The current intensity of this new reduction peak increases linearly with γ (inset, Fig. 4a) and shifts gradually toward more negative potentials (γ = 7, Epc = − 1.118 V vs Ag/AgCl; γ = 8, Epc = − 1.184 V vs Ag/AgCl; γ = 9, Epc = − 1.218 V vs Ag/AgCl; γ = 10, Epc = − 1.267 V vs Ag/AgCl). Observably, this reduction process is accompanied by the formation of bubbles on the surface of the electrode for γ values above 9, assigning this wave to an electrocatalytic behavior for the reduction of protons and evolution of dihydrogen gas [14, 37,38,39]. This was confirmed via bulk electrolysis experiments and gas chromatography tests definitely confirming that hydrogen gas is produced at the working electrode. As a matter of fact, the newly formed wave in the presence of acid does not modify the current intensities and potential locations of the voltammetric pattern associated with compound 1 in the absence of acid.

Cyclic voltammograms of the compounds 1 (a), 2 (b) and 3 (c) in DMF + 0.1 M Bu4NPF6 with stepwise addition of TFA. The arrow indicates the increasing values of the excess parameter γ = [acid]/[POTM] (below 10). Comparison of the cyclic voltammograms for the blank and compounds 1 − 3 (d) at γ = 10. Scan rate was 100 mV s−1
Likewise, the addition of increasing amounts of TFA in the solution of compound 2 or 3 also leads to the growing of a new composite cathodic wave when γ increase to 6 or 8, respectively. Such a catalytic wave is not observed at bare glassy carbon electrodes at γ = 10 in the absence of compounds 1 − 3, which demonstrate HER catalytic behavior of compounds 1 − 3 (Fig. 4d). The CVs recorded at γ = 8 highlight some discrepancies between three compounds. At γ = 8, the new cathodic peak potentials measured for compound 1 is found to be − 1.184 V vs. Ag/AgCl and move to − 1.282 V vs. Ag/AgCl for compound 2 and to − 1.208 V vs. Ag/AgCl for compound 3, respectively. This indicates that compound 1 is more prone to proton reduction at more positive potential than compound 2 or 3, probably because compound 1 has more [Mo2O2S2] units. Despite more negative proton reduction potential, compound 2 has a larger peak current than compound 1 or 3 probably due to the ligand anchored inside Mo-ring.
In terms of the catalytic active components, the bridging sulfides were proved to be essential in the catalytic process, which act as stabilizing agents for the reduction of Mo centers. The electrochemical studies for Mo8S8(Ale)4 reported by H. El Moll [16] demonstrate unambiguously that only the sulfurated compound is active for the reduction of protons into hydrogen while its oxo analogue does not exhibit any electrocatalytic behavior. Besenbacher et al. suggested that edge sulfur atoms could be viewed as a significant fraction of active sites [40]. Hijazi et al. reported that the carboxylate ligands encapsulated within the Mo-ring probably play the role of proton relay and may be protonated in the first step of the catalytic process [28]. Carboxylic acid ligands can be used as templates to obtain such cyclic clusters, but their effects on the electrocatalytic properties of the compounds are not significant. Therefore, the discrepancies in HER electrocatalytic process of compounds 1 − 3 evidence the important role played by the [Mo2O2S2] units. This family of oxothiomolybdenum cycles appears as efficient electro-catalysts in organic medium and prompts us to develop the study of these compounds. Further experiments are in progress to investigate deeply the electro-catalytic functioning of these Mo–S cyclic materials.
Conclusions
Three cyclic polyoxothiomolybdates encapsulating different dicarboxylates, namely [Mo20-(PyD)2]4−, [Mo12-NapH]2− and [Mo8-Ox]2− were synthesized and characterized in the solid state and in solution and these compounds were studied by X-ray diffraction on single crystals. The electrochemical studies revealed that this class of cyclic compounds act as efficient electro-catalysts for the reduction of protons into hydrogen in organic medium. The possibility of accumulating properties at the molecular level in such multicomponent systems offers promising prospects for fine-tuning the electrochemical reactivity of these preliminary results.
References
A. Seko, T. Yamase, and K. Yamashita (2009). J. Inorg. Biochem. 103, 1061.
A. Ogata, H. Yanagie, E. Ishikawa, Y. Morishita, S. Mitsui, A. Yamashita, K. Hasumi, S. Takamoto, T. Yamase, and M. Eriguchi (2008). Br. J. Cancer. 98, 399.
A. Giusti, G. Charron, S. Mazerat, J. D. Compain, P. Mialane, A. Dolbecq, E. Riviere, W. Wernsdorfer, B. R. Ngo, B. Keita, L. Nadjo, A. Filoramo, J. P. Bourgoin, and T. Mallah (2009). Angew. Chem. Int. Ed. 48, 4949.
J. Compain, P. Mialane, A. Dolbecq, I. M. Mbomekallé, J. Marrot, F. Sécheresse, E. Rivière, G. Rogez, and W. Wernsdorfer (2009). Angew. Chem. Int. Ed. 48, 3077.
E. Coronado, C. Giménez-Saiz, and C. J. Gómez-García (2005). Coord. Chem. Rev. 249, 1776.
A. Dolbecq, C. Peloux, A. L. Auberty, S. A. Mason, P. Barboux, J. Marrot, E. Cadot, and F. Sécheresse (2002). Chem. Eur. J. 8, 349.
J. Yan, J. Gao, D. L. Long, H. N. Miras, and L. Cronin (2010). J. Am. Chem. Soc. 132, 11410.
H. N. Miras, G. J. Cooper, D. L. Long, H. Bögge, A. Müller, C. Streb, and L. Cronin (2010). Science. 327, 72.
A. M. Appel, D. L. DuBois and M. Rakowski DuBois (2005). J. Am. Chem. Soc. 127, 12717.
L. L. Lopez, P. Bernatis, J. Birnbaum, R. C. Hattiwanger, and M. Rakowski DuBois (1992). Organometallics. 11, 2424.
J. C. V. Laurie, L. Duncan, R. C. Haltiwanger, R. T. Weberg, and M. Rakowski DuBois (1987). ChemInform. 18, 6234.
A. Miller, H. Dornfeld, G. Henkel, B. Krebs, and M. P. A. Viegers (1978). Angew. Chem. Int. Ed. 17, 52.
S. Duval, S. Floquet, C. Simonnet-Jégat, J. Marrot, R. N. Biboum, B. Keita, L. Nadjo, M. Haouas, F. Taulelle, and E. Cadot (2010). J. Am. Chem. Soc. 132, 2069.
B. Keita, S. Floquet, J. Lemonnier, E. Cadot, A. Kachmar, M. Bénard, M. Rohmer, and L. Nadjo (2008). J. Phys. Chem. C. 112, 1109.
A. Dolbecq, E. Cadot, and F. Sécheresse (2000). Comptes Rendus De Lacademie Des Sciences. 3, 193.
B. Salignac, S. Riedel, A. Dolbecq, F. Sécheresse, and E. Cadot (2000). J. Am. Chem. Soc. 122, 10381.
E. Cadot, J. Marrot, and F. Sécheresse (2001). Angew. Chem. Int. Ed. 40, 774.
S. Floquet, J. Marrot, and E. Cadot (2005). CR. Chem. 8, 1067.
J. Lemonnier, S. Floquet, A. Kachmar, M. Rohmer, M. Bénard, J. Marrot, E. Terazzi, C. Piguet, and E. Cadot (2007). Dalton Trans. 28, 3043.
J. Lemonnier, S. Floquet, J. Marrot, E. Terazzi, C. Piguet, P. Lesot, A. Pinto, and E. Cadot (2007). Chem. Eur. J. 13, 3548.
J. F. Lemonnier, A. Kachmar, S. Floquet, J. Marrot, M. M. Rohmer, M. Benard, and E. Cadot (2008). Dalton Trans. 34, 4565.
A. Kachmar, S. Floquet, J. Lemonnier, E. Cadot, M. Rohmer, and M. Bénard (2009). Inorg. Chem. 48, 6852.
J. W. Purcell, D. L. Long, E. C. Lee, L. Cronin, and H. N. Miras (2018). Dalton Trans. 47, 6283.
J. W. Purcell, H. N. Miras, D. L. Long, P. Markopoulou, and L. Cronin (2017). Chem. Eur. J. 23, 9683.
H. Y. Zang, A. Surman, D. L. Long, L. Cronin, and H. N. Miras (2016). Chem. Commun. 52, 9109.
A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, C. R. Mayer, V. Artero, and E. Cadot (2011). Inorg. Chem. 50, 9031.
H. El Moll, J. C. Kemmegne-Mbouguen, M. Haouas, F. Taulelle, J. Marrot, E. Cadot, P. Mialane, S. Floquet, and A. Dolbecq (2012). Dalton Trans. 41, 9955.
E. Cadot, B. Salignac, S. Halut, and F. Sécheresse (1998). Angew. Chem. Int. Ed. 37, 611.
V. Béreau, E. Cadot, H. Bögge, A. Müller, and F. Sécheresse (1999). Inorg. Chem. 38, 5803.
A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, J. Fize, V. Artero, O. David, E. Magnier, B. Pégot, and E. Cadot (2013). Dalton Trans. 42, 4848.
S. Floquet, S. Draoui, J. Marrot, F. Millange, M. Frigoli, and E. Cadot (2014). J. Cluster Sci. 25, 811.
D. Li, Y. Xing, Z. Li, J. Xu, W. Song, T. Wang, G. Yang, N. Hu, H. Jia, and H. Zhang (2005). J. Inorg. Biochem. 99, 1602.
H. Zang, H. N. Miras, J. Yan, D. L. Long, and L. Cronin (2012). J. Am. Chem. Soc. 134, 11376.
H. Zang, A. R. de la Oliva, H. N. Miras, D. Long, R. T. McBurney, and L. Cronin (2014). Nat. Commun. 5, 3715.
H. Y. Zang, H. N. Miras, D. L. Long, B. Rausch, and L. Cronin (2013). Angew. Chem. Int. Ed. 52, 6903.
Y. L. Zhong, W. Ng, J. Yang, and K. P. Loh (2009). J. Am. Chem. Soc. 131, 18293.
Y. Oudart, V. Artero, J. Pécaut, and M. Fontecave (2006). Inorg. Chem. 45, 4334.
R. Angamuthu and E. Bouwman (2009). Phys. Chem. Chem. Phys. 11, 5578.
A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin, and M. Fontecave (2009). Science. 326, 1384.
J. Kibsgaard, T. F. Jaramillo, and F. Besenbacher (2014). Nat. Chem. 6, 248.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21471003 and 21372007); Natural Science Foundation of Educational Commission of Anhui Province of China (No. KJ2020A0240).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, QY., Xin, Z., Hu, MJ. et al. Syntheses, Characterizations and Properties of [Mo2O2S2]-Based Oxothiomolybdenum Rings Incorporating Carboxylate Ligands. J Clust Sci 34, 285–295 (2023). https://doi.org/10.1007/s10876-022-02219-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-022-02219-4