
Abstract
The geometric structures and properties of mixed silicon/gold clusters (Si n Au2; n = 1–10) are theoretically studied using density functional theory and coupled cluster calculations. Several new low lying isomers are found for most cluster sizes. New lowest energy isomers are found for Si7Au2, Si9Au2, and Si10Au2. Si1Au2 and Si3Au2 are predicted to be planar structures, whereas other sizes are three dimensional. Energetically, Si5Au2 and Si8Au2 are predicted to be more stable clusters compared to other sizes investigated here. For all lowest energy structures, gold atoms are bound exohedrally on the cluster and the frontier orbitals often show interactions between silicon p and gold d atomic orbitals. Only for the largest sized cluster, Si10Au2, does the cluster take the same structure of the pure Si10 cluster with Au atoms capping it. For other cluster sizes, at least one Au atom makes up part of the central structural motif. Natural bond order analysis is performed to further explore the bonding in each cluster.
Graphical Abstract
Silicon clusters doped with two gold atoms, Si n Au2 (n = 1–10) are studied by density functional and coupled cluster calculations. Pictured is the lowest energy isomer of Si7Au2, one of the new novel ground state structures reported here, and the calculated total density surface.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Silicon is one of the most important elements to the electronics industry due to its semiconducting properties. The precise structures, and consequently the properties, of silicon clusters can be varied by introducing different dopant atoms. For example, in industry doping silicon is important to its use in transistors, solar cells, and other technologies. Additionally, coinage metals can act as catalysts in the growth is silicon nanowires, where gold is preferred over silver and copper due to the lower eutectic temperature of the Si–Au mixture [1, 2]. Recently, it has been shown that the most probable doping site for the metal is on the surface of nano sized silicon crystals, and that gold atoms on the surface then promote more Si–Si covalent bonding compared to silver and copper which introduce some ionicity [2]. It has also been demonstrated that a single gold dopant atom may start to occupy interior sites in silicon clusters for Si11 and larger clusters, and that Au@Si12 may possess some enhanced chemical activity [3].
Pure gold clusters show several unique properties as well. For example, small gold clusters possess 2D structures, but larger clusters have 3D structures [4–6]. In addition, the catalytic ability of each cluster depends on many factors, such as the size and shape of the clusters [7, 8]. Doping gold clusters with silicon atoms is another route to control the catalytic ability of gold clusters. For example, a doped Au7Si cluster has the ability to activate molecular oxygen, whereas a pure gold cluster with eight atoms shows no catalytic activity [9]. Au4Si has a highly stable geometry, exhibiting strong Si–Au bonding [10]. Doping small gold cluster anions with a silicon atom have shown a change from 3D to quasi-2D to 3D structures when going from SiAu -5 to SiAu8 − [11], and doping gold clusters with silicon atoms have demonstrated an analogy between gold and hydrogen [12–15]. Theoretical predictions of larger gold clusters doped with a single silicon atom (SiAu n ; n = 17–20) have shown how the cluster structures are significantly different compared to pure gold clusters [16]. The structures of pure gold clusters are also very different from pure silicon clusters. For example, Au n (n = 6–8) are two dimensional planar structures, whereas silicon clusters of the same size are 3D structures [4, 17]. This leads one to wonder at what silicon/gold concentration a cluster will resemble the structure of a pure atomic cluster.
Despite the number of studies focusing on silicon clusters doped with a single metal atom, only a select few investigations have looked at silicon clusters doped with two transition metal atoms. These theoretical studies include different sized silicon clusters doped with two transition metal atoms of Mo [18], V [19], Cr [20], Mn [20], Fe [21], Co [21], Ni [21], Cu [22], Ag [23], and Au [24]. There have also been only a few experimental studies on these doubly doped silicon clusters. For example, anion photoelectron spectra have been collected for a single sized V2Si20 cluster [19], and mass spectra of doubly 3d transition metal doped silicon cluster cations have determined at what size the dopant changes from being exohedrally bound to endohedral in the silicon cluster [25]. Introducing two dopant metal atoms may lead to a competition between M–Si and M–M bonding in the cluster [26]. Although small Si n Au2 (n = 1–8) clusters have been theoretically studied previously [24], further investigations and analysis warrant consideration. Here, we report several new low lying isomers, a new ground state structure for n = 7, and extend the upper limit of the size range studied from Si8Au2 to Si10Au2. The new ground state structure of Si7Au2 changes the predicted stabilization energies of Si6Au2 and Si8Au2. We then further study the properties of the clusters by analyzing the frontier molecular orbitals of the clusters and performing Natural Bond Order (NBO) calculations. Lastly, detailed coupled cluster calculations are performed on small sizes Si n Au2 (n = 1–3) for comparison.
Theoretical Methods
Cluster structure calculations were performed using the Gaussian’09 program [27]. Computational time for this project was awarded through the eXtreme Science and Engineering Discovery Environment (XSEDE) [28]. The BP86 density functional method was employed with the SVP basis set for Si atoms and the SDD pseudopotential on Au atoms [29–32]. This method was utilized here as it was previously shown to accurately reproduce experimental infrared spectra for the lowest energy structural isomers of similar systems such as pure silicon clusters [33–35], as well as well as silicon clusters doped with a single silver or gold atom [36, 37]. In addition, this 19 valence electron effective core potential on gold, has been shown in other studies to be suitable in modeling weak Au–Au interactions, which may be present in the clusters studied here with two gold atoms [38]. Initial structures were taken from predicted low energy structures of related systems [18, 20–24, 37], adding or removing atoms from calculations on different sized clusters, or from considering new cluster structure orientations. Cluster structures were optimized without symmetry constraints and contain all real frequencies. Reported relative energy values include zero-point energy corrections. Calculations were performed with both singlet and triplet spin states. For all lowest energy isomers, a spin multiplicity of one was found to be more favorable. Graphical representations of molecular orbitals were created with the cubegen utility, and natural atomic and bond analysis was completed with NBO version 6.0 [39]. For select small sized Si n Au2 clusters, the Coupled Cluster Single and Double excitation (CCSD) method [40] was also employed with the cc-pVTZ basis on silicon atoms [41] and the SDD pseudopotential on Au [32]. The results from these calculations, which also include zero-point corrections to the energy, are used for comparison with our BP86 results, and are discussed in a latter section of this article.
Results and Discussion
Geometric Structures
Si n Au2 (n = 1–5)
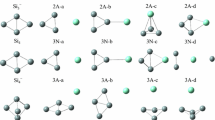
The predicted ground state and low energy isomers for Si n Au2 (n = 1–5) are shown in Fig. 1. The ground state structures in this size range are the same as predicted previously with a different DFT method [24]. Here, we discuss these ground state structures as well as new low energy isomers explored in this investigation. As experimentally determined structures of similar Si n Au+ have been reported, we also compare our lowest energy isomers with those geometries.

Optimized geometry of ground state and low energy Si n Au2 (n = 1–5) isomers. Relative energy values reported for each isomer are in kJ/mol
The lowest energy Si1Au2 isomer (SiAu2-a) is predicted to be a bent structure with a central silicon atom. The predicted Si–Au bond length (2.309 Å) is elongated from that predicted for the SiAu dimer (2.286 Å). This structure has been predicted previously to be the ground state structure of neutral SiAu2 by different DFT methods [10, 24], and is similar to the experimentally determined Si2Au+ structure, which is assigned to an isosceles triangle [37].
Si2Au2 is predicted to have a dibridged structure (isomer Si2Au2-a), which is the structure that is experimentally known for anionic Si2Au2 − from photoelectron spectroscopy [13]. No other isomer is close in energy, with the next structure (isomer Si2Au2-b) lying over 39 kJ/mol higher in energy. The Si–Au bond length is longer (2.505 Å) than that of the smaller SiAu2 cluster, and the predicted Si–Si bond length (2.398 Å) is similar to the bond lengths predicted for pure silicon clusters. Cationic Si3Au+, which also contains four Si and Au atoms, was previously assigned to a planar rhombus structure based on the observed infrared spectra [37]. A similar rhombic Si2Au2 structure (isomer Si2Au2-c) is predicted here to be 116.9 kJ/mol higher in energy than the lowest energy dibridged structure.
The Si3Au2 cluster is calculated to be planar, where a gold atom caps the apex of a slightly distorted square planar geometry (isomer Si3Au2-a). This is similar to the structure assigned for the five atom Si4Au+ cluster, where the gold atom binds to the apex of a silicon rhombus [37]. The capping Au atom is predicted to have a bond length of 2.296 Å with a silicon atom, whereas the Au atom that is part of the square structure has longer bond lengths of 2.385 Å with the silicon atoms. Here, the second and third lowest energy isomers (isomers Si3Au2-b and Si3Au2-c, respectively) are predicted with our BP86 method in the opposite order from that reported previously using a different DFT method [24], but both methods predict the two structural isomers to be relatively close in energy. Our CCSD method calculations also predicts that Si3Au2-b is lower in energy than Si3Au2-c (see section on CCSD results below), suggesting that this is the correct energetic ordering of these isomers. The low energy structure Si3Au2-e is a new structural isomers not previously considered.
Calculations predict Si4Au2 to have a geometry resembling that of Si2Au2, with two additional silicon atoms capping the top of the structure (isomer Si4Au2-a). Here the average Au–Si bond length (2.511 Å) is only slightly larger than that of the predicted ground state Si2Au2 structure. Rotating this structure, it can also be seen as a distorted trigonal prism. Isomers Si4Au2-b and Si4Au2-e are new structures not previously reported. The new isomer Si4Au2-b, which is a distorted tirgional prism, is almost identical in energy with the predicted ground structure, lying only 0.7 kJ/mol higher in energy.
The predicted structure for Si5Au2 resembles a cubic structure with one of the corner atoms removed (isomer Si5Au2-a). This ground state structure has the smallest Au–Au distances (3.220 Å) of any of the ground state structures for all cluster sizes in this investigation. The structure is slightly lower in energy (7.4 kJ/mol) than the second lowest energy isomer, a capped trigional prism structure (isomer Si5Au2-b). Si6Au+ has been previously assigned to a structure where the gold atom binds to a distorted silicon octahedron [37]. Calculations on a similar shaped Si5Au2 structure (Si5Au2-g) was predicted here to be 42.9 kJ/mol higher in energy than the lowest energy Si5Au2-a cluster. Of the isomers displayed in Fig. 1, only Si5Au2-a and Si5Au2-c have been previously reported [24].
Si n Au2 (n = 6–8)
The ground state and low energy structural isomers of Si n Au2 in this size range are shown in Fig. 2. The lowest energy isomer for Si6Au2 can be viewed as a distorted diamond or prism structure (isomer Si6Au2-a), which is related to the Si5Au2 ground state structure with an additional Si atom adding to the base plane. Changing the locations of a single Si and Au atom raises the energy 5.7 kJ/mol to the second lowest energy isomer (Si6Au2-b).

Optimized geometry of ground state and low energy Si n Au2 (n = 6–8) isomers. Relative energy values reported for each isomer are in kJ/mol
Si7Au2 is predicted to be a new novel structure which has not been predicted previously, with a trigional prism structural motif and additional face and edge caps (isomer Si7Au2-a). This isomer lies only slightly lower in energy (3.4 kJ/mol) to isomer Si7Au2-b, which mainly differs by the position of one of the Au atoms. The lowest energy isomer is dramatically different from the experimentally determined Si8Au+ structure, which contains an edge capped pentagonal bipyrimidal silicon structural motif with the Au atom bound to a Si–Si edge [37]. The previously predicted lowest energy structure for this cluster size (isomer Si7Au2-g) lies over 40 kJ/mol higher in energy [24], and several other structures (isomers Si7Au2-a through Si7Au2-f in Fig. 2) are found that lie lower in energy compared to this structure. When we performed calculations on these structures at the theoretical level previous used by Zhou, et al. [24] (TPSS functional, 6-311 + G(d) basis for Si and LANL2TZ(f) effective core potential for Au [42–44]), Si7Au2-b is the lowest energy isomer, lying only 0.4 kJ/mol lower in energy than Si7Au2-a. Both isomers are considerably more favorable than the previously reported lowest energy isomer Si7Au2-g (which is 34.2 kJ/mol higher in energy than Si7Au2-b at this level off theory). The six isomers predicted to be lower in energy than Si7Au2-g in Fig. 2 are all also predicted to be lower in energy with the TPSS method as well.
The geometry of Si8Au2 is a drum like pentagonal structure (isomer Si8Au2-a). This is a different structure from the experimentally determined Si9Au+ structure, which is based on a pentagonal bipyrimidal structural motif [37]. Several new stable structural isomers are found and investigated here, but none are close in energy to Si8Au2-a, with the closest being over 29 kJ/mol higher in energy. The structure of Si8Au2-a resembles a low energy isomer (i.e., Si7Au2-c) of the previous cluster size.
Si n Au2 (n = 9, 10)
Here, we extend previous investigations on Si n Au2 clusters to larger sizes which have not been studied previously. The structures in this size range are shown in Fig. 3. Additional higher energy isomers are presented in the supporting information. The ground state structure of Si9Au2 (isomer Si9Au2-a) has a structure similar to Si8Au2-a, with a sixth Si atom inserted into the previous five membered ring of silicon atoms. The structure has a short Au–Au distance (3.319 Å), only slightly longer than the Au–Au distances in Si5Au2-a (3.220 Å) and Si2Au2-a (3.274 Å). The second lowest energy isomer, Si9Au2-b, is nearly 20 kJ/mol higher in energy. Si9Au2-c and Si9Au2-d are essentially identical structures with differing positions of an Au atom.

Optimized geometry of ground state and low energy Si n Au2 (n = 9, 10) isomers. Relative energy values reported for each isomer are in kJ/mol. Additional higher energy isomers are presented in the supporting information
The lowest energy Si10Au2 isomer is predicted to have a tricapped trigonal prism motif, with an extra silicon cap as well as the two exohedral gold capping atoms (isomer Si10Au2-a). The tricapped trigonal prism structural motif is also a common shape for pure silicon clusters [33–35]. There are three additional low energy structural isomers that are all within 7 kJ/mol of the ground state structure, and they all contain a trigional prism motif. It appears that at this size, the mixed clusters have a large enough silicon ratio to have a ground state structure similar to pure silicon clusters with the two dopant Au atoms binding exohedrally to outside locations of the cluster. This is different from smaller sized Si n Au2 clusters where at least one Au atom is a part of the main structural motif. Indeed, the silicon framework of this lowest energy isomer is almost exactly the same as that for Si10 as assigned from experimental IR-MPD spectra [35], with one cap being moved slightly to allow for the presence of one of the Au atoms. This ground state structure has the largest distance between the two Au atoms (7.269 Å) of all of the lowest energy structures in the size range investigated here.
Comparison of DFT Method and Coupled Cluster
In order to further the validity of the chosen density functional theoretical method employed for this investigation, coupled cluster calculations using both single and double substitutions (i.e., CCSD) with the cc-pVTZ basis set for Si and the SDD pseudopotential for Au atoms were performed for small sized clusters. For SiAu2, CCSD calculations predict a similar ground state structure to the BP86 method, with a slightly shorter Si–Au bond (2.264 Å for CCSD vs. 2.309 Å for BP86). Similar to the BP86 calculations, CCSD calculations also predict Si2Au2-a to be the lowest energy isomer, lying 43.9 kJ/mol lower than Si2Au2-b and 129.0 kJ/mol lower than Si2Au2-c. These relative energy values are in good agreement with that predicted by our BP86 calculations here (39.8 and 116.9 kJ/mol, respectively). The predicted geometry of Si2Au2-a is similar between the CCSD method (with 2.464 Å Si–Au bond lengths and a 2.333 Å Si–Si bond length) and BP86 method (with 2.505 Å Si–Au bond lengths and a 2.398 Å Si–Si bond length). Si3Au2-a was also predicted to be the ground electronic state of this sized cluster at the CCSD level of theory. Similar to our earlier DFT results, Si3Au2-b and Si3Au2-c are predicted to be only slightly more unstable compared to Si3Au2-a, lying 5.0 and 6.2 kJ/mol higher in energy with the CCSD method, respectively. This shows that our BP86 calculations, which predicts Si3Au2-b and Si3Au2-c to be 11.9 and 14.1 kJ/mol higher than Si3Au2-a, predict the relative energy of the Si3Au2 isomers in the same order as the CCSD method, compared to previous TPSS calculations which predict Si3Au2-c to be lower in energy than Si3Au2-b. These CCSD results for small sized Si n Au2 clusters are in good agreement with our BP86 results for these sizes, and further validates the BP86 functional as an appropriate method to predict the electronic and structural properties of larger clusters. Earlier results have already successfully utilized the BP86 method to predict the electronic and spectroscopic properties of similar Si n and Si n M+ (M = Ag and Au) clusters [33–37].
Structural Properties and Trends
Figure 4 shows the atomic binding energy and fragmentation energy, calculated in electron volts. The numerical values of the binding energy are given in Table 1. The atomic binding energy is calculated as

The atomic binding energy (black) and dissociation energy (red) for predicted Si n Au2 (n = 1–10) clusters calculated in electron volts. Peaks at Si5Au2 and Si8Au2 indicate enhanced stability. See text for details on how each is calculated
and the fragmentation energy is calculated as
The binding energy shows a smooth increase, with small bumps for Si5Au2 and Si8Au2. The fragmentation energy also identifies an enhanced stability as maxima for these two cluster sizes. This is confirmed as minima (largest negative numbers) when the relative stability of each cluster sizes, shown in Fig. 5, is calculated as

Relative stabilization energy, calculated in electron volts, for Si n Au2 (n = 1–9). Minima for Si5Au2 and Si8Au2 indicate cluster sizes with enhanced stability relative to clusters with one less and one more Si atoms. See text for details on how numerical values are calculated
Looking at the ground state cluster structures as a function of cluster size, one notices that Si1Au2 is planar, Si2Au2 is three dimensional, Si3Au2 is again 2D, and larger clusters are all 3D. It is interesting that Si3Au2 is 2D, whereas Si2Au2 is 3D. This is, perhaps, due to the fact that Si2Au2 is composed equally of silicon and gold atoms, whereas Si3Au2 and larger clusters are predominantly composed of silicon atoms doped with 2 gold atoms. Indeed, small mixed silicon gold clusters that are predominantly gold (Si n Au m ; n = 1, 2, m = 2–4) have been shown to possess unusual structures similar to silicon hydrides [12–15]. This possibly suggests the structures of larger Si n Au2 clusters could differ drastically if the Au concentration is further increased.
The Natural Electronic Configuration (NEC) for the two gold atoms in the lowest energy structure for each cluster size is given in Table 1. All of the gold atoms have <10 electrons in the 5d orbital, indicating that the atoms have donated electron density from this atomic orbital when bonding. Although there are some exceptions for larger clusters, in general from SiAu2 through Si5Au2 it appears the number of electrons in the 6 s decreases, and the number of electrons in the 6p increases as the cluster size increases. This indicates that gold atoms in very small Si n Au2 clusters prefer to accept electron density into the 6s atomic orbital when binding whereas gold atoms in larger cluster accept electron density in the 6p orbital. The NEC for silicon atoms listed in the same table show that the Si atoms in the cluster donate electron density from their 3s atomic orbitals, and accept density into their 3p orbitals. The exception is for the smallest size SiAu2 cluster where the Si atom donates from both its 3s and 3p orbitals.
Natural Resonance Theory (NRT) analysis was performed for the smallest sized SiAu2 and Si2Au2 clusters. For SiAu2, a total natural bond order of 1.50 is predicted between the Si atom and each Au atom. In Si2Au2, the total natural bond order is 0.53 between each Si and Au atom, and the natural bond order between the two Si atoms is predicted to be 0.98. For both sized clusters NRT analysis predicts the total Si–Au bonding to be made up approximately half from covalent bond order and half from ionic bond order, whereas the total Si–Si bond order is composed almost entirely from covalent interactions in Si2Au2. For both sized clusters, the Au–Au total natural bond order is 0.00, and bonding interactions are not predicted to be significant.
In inspecting the bond character from the NBO calculations on the lowest energy isomers, in SiAu2, the Si–Au bonds are predominantly made from the s orbital of Au (89.8 %) and the p orbital of Si (95.7 %). In Si2Au2, the Si–Au bonds are similar (96.7 % Au s atomic orbital and 98.0 % Si p character). The Si–Si bond is composed from 88 % or the p atomic orbital on both Si. In all larger clusters, the results are similar. That is, the bond order between Si and Au is predominately composed of the s orbital on Au and the p orbital of Si.
Mullikan (QMull) and Natural (QNat) changes for the two gold atoms in each sized cluster are presented in Table 1. Several studies have suggested that the natural changes from NBO analysis is more reliable than other methods at predicting the charge compared to experimental results [45–47]. Mullikan charges are given for comparison, as they are one the most widely used charge schemes. The two methods seem to agree reasonable well for the charge of the gold atoms in SiAu2. Beyond that, however, natural charges predict that the gold atoms will have an overall positive charge, whereas Mullikan charge predict the gold atoms will have a negative charge (except for Si10Au2 where one gold atoms is predicted to have a positive Mullikan charge). The natural charges also predict that the overall gold charge becomes increasingly positive with cluster size from Si1Au2 to Si5Au2, and is most positive for Si8Au2 and Si9Au2.
The frontier orbitals (i.e., HOMO and LUMO) for the lowest energy structure for each cluster size are shown in Fig. 6, and several things can be commented on. First, the HOMO of Si2Au2 appears to be mostly a bonding overlap between silicon p atomic orbitals. The LUMO also contains a bonding interaction between silicon p orbitals. Hence, if this is also the ground state structure of ionic Si2Au2 + and Si2Au2 −, one could expect the Si–Si bond to be strongest for the anion and weakest for the cation. Likewise, the HOMO or Si3Au2 is mainly a bonding interaction between p atomic orbitals of all three silicon atoms. The frontier orbitals of larger clusters become increasingly complicated. However, one can clearly discern silicon p orbitals and mainly gold d orbitals participating in forming the molecular orbitals through bonding, nonbonding, and antibonding interactions. For example, the HOMO of Si9Au2 appears to be constructed from silicon p and gold d 2z atomic orbitals.

Highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO) of the predicted lowest energy Si n Au2 (n = 1–10) clusters
Conclusion
Silicon clusters doped with two gold atoms (Si n Au2, n = 1–10) are theoretically studied with density functional and coupled cluster methods. It is found that SiAu2 and Si3Au2 prefer a 2D structure, whereas larger cluster sizes are 3D. Several new low energy isomers are found, including a new ground state structure for Si7Au2. We also investigate larger Si9Au2 and Si10Au2 clusters which have not been previously studied. Si10Au2 is found to be the first cluster size where both gold atoms cap a bare Si10 cluster, whereas smaller clusters incorporate at least one Au atom in the base structural motif. Si5Au2 and Si8Au2 are clusters with enhanced stability compared to other sizes. NBO analysis indicates the gold atoms for all ground state clusters except SiAu2 have a net positive charge, and analysis of the frontier orbitals indicate they are predominantly constructed from the mixing silicon p and gold d atomic orbitals.
References
D. Parlevliet and J. C. L. Cornish (2011). MRS Proc. 989, 0989.
C. L. Mayfield and M. N. Huda (2014). Chem. Phys. Lett. 605–606, 38.
J. Wang, Y. Liu, and Y.-C. Li (2010). Phys. Lett. A 374, 2736.
P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer, and A. Fielicke (2008). Science 321, 674.
M. P. Johansson, I. Warnke, A. Le, and F. Furche (2014). J. Phys. Chem. C 118, 29370.
J. A. Hansen, P. Piecuch, and B. G. Levine (2013). J. Chem. Phys. 139, 091101.
B. Yoon, H. Häkkinen, and U. Landman (2003). J. Phys. Chem. A 107, 4066.
R. Pal, L.-M. Wang, Y. Pei, L.-S. Wang, and X. C. Zeng (2012). J. Am. Chem. Soc. 134, 9438.
D. Manzoor, S. Krishnamurty, and S. Pal (2014). J. Phys. Chem. C 118, 7501.
M. Zhang, S.-B. Yang, X.-J. Feng, L.-X. Zhao, H.-Y. Xhang, and Y.-H. Luo (2013). Eur. Phys. J. D 67, 11.
R. Pal, L.-M. Wang, W. Huang, L.-S. Wang, and X. C. Zeng (2009). J. Am. Chem. Soc. 131, 3396.
B. Kiran, X. Li, H.-J. Zhai, L. F. Cui, and L. S. Wang (2004). Angew. Chem. Int. Ed. 43, 2125.
X. Li, B. Kiran, and L.-W. Wang (2005). J. Phys. Chem. A 109, 4366.
B. Kiran, X. Li, H.-J. Zhai, and L.-S. Wang (2006). J. Chem. Phys. 125, 133204.
L.-S. Wang (2010). Phys. Chem. Chem. Phys. 12, 8694.
H.-W. Yang, W.-C. Lu, L.-Z. Zhao, W. Qin, W.-H. Yang, and X.-Y. Xue (2013). J. Phys. Chem. A 117, 2672.
P. Gruene, B. Butschke, J. T. Lyon, D. M. Rayner, and A. Fielicke (2014). Z. Phys. Chem. 228, 337.
J.-G. Han, R.-N. Zhao, and Y. Duan (2007). J. Phys. Chem. A 111, 2148.
H.-G. Xu, X.-Y. Kong, X.-J. Deng, Z.-G. Zhang, and W.-J. Zheng (2014). J. Chem. Phys. 140, 024308.
R. Robles, S. N. Khanna, and A. W. Castleman Jr (2008). Phys. Rev. B 77, 235441.
R. Robles and S. N. Khanna (2009). J. Chem. Phys. 130, 164313.
P. Shao, X.-Y. Kuang, L.-P. Ding, M.-M. Zhong, and Z.-H. Wang (2012). Physica B 407, 4379.
Y.-R. Zhao, X.-Y. Kuang, S.-J. Wang, Y.-F. Li, and P. Lu (2011). Z. Naturforsch., A Phys. Sci. 66, 353.
Z.-J. Zhou and Y.-F. Hu (2012). Z. Naturforsch. 67a, 99.
E. Janssens, P. Gruene, G. Meijer, L. Wöste, P. Lievens, and A. Fielicke (2007). Phys. Rev. Lett. 99, 063401.
Y. Cao, C. van der Linde, R. F. Höckendorf, and M. K. Beyer (2010). J. Chem. Phys. 132, 224307.
Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka, G. D. Peterson, R. Roskies, J. R. Scott, and N. Wilkins-Diehr (2014). Comput. Sci. Eng. 16, (5), 62.
A. D. Becke (1988). Phys. Rev. A 38, 3098.
J. P. Perdew (1986). Phys. Rev. B 33, 8822.
A. Schaefer, H. Horn, and R. Ahlrichs (1992). J. Chem. Phys. 97, 2571.
D. Andrae, U. Haeussermann, M. Dolg, H. Stoll, and H. Preuss (1990). Theor. Chem. Acc. 77, 123.
J. T. Lyon, P. Gruene, G. Meijer, A. Fielicke, E. Janssens, P. Claes, and P. Lievens (2009). J. Am. Chem. Soc. 131, 1115.
A. Fielicke, J. T. Lyon, M. Härtelt, G. Meijer, P. Claes, J. de Haeck, and P. Lievens (2009). J. Chem. Phys. 131, 171105.
M. Haertelt, J. T. Lyon, P. Claes, J. de Haeck, P. Lievens, and A. Fielicke (2012). J. Chem. Phys. 136, 064301.
Y. Li, J. T. Lyon, A. P. Woodham, A. Fielicke, and E. Janssens (2014). ChemPhysChem 15, 328.
Y. Li, J. T. Lyon, A. P. Woodham, P. Lievens, A. Fielicke, and E. Janssens (2015). J. Phys. Chem. C 119, 10896.
P. Zaleski-Ejgierd and P. Pyykkö (2009). Can. J. Chem. 87, 798.
NBO 6.0. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, and F. Weinhold (Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013). http://nbo6.chem.wisc.edu/
G. E. Scuseria, C. L. Janssen, and H. F. Schaefer III (1988). J. Chem. Phys. 89, 7382.
D. E. Woon and T. H. Dunning Jr (1993). J. Chem. Phys. 98, 1358.
J. M. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria (2003). Phys. Rev. Lett. 91, 146401.
A. D. McLean and G. S. Chandler (1980). J. Chem. Phys. 72, 5639.
P. J. Hay and W. R. Wadt (1985). J. Chem. Phys. 82, 270.
R. Yerushalmi, A. Scherz, and K. K. Baldridge (2004). J. Am. Chem. Soc. 126, 5897.
K. C. Gross, P. G. Seybold, and C. M. Hadad (2002). Int. J. Quantum Chem. 90, 445.
J. B. Levy (1999). Struct. Chem. 10, 121.
Acknowledgments
Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research. This work used the eXtreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. Acknowledgment is also given for support from Clayton State University through the University Undergraduate Research and Creative Activities (URCA) and the College of Arts and Sciences’ Mini Grants and Creative Activities and Scholarship Enrichment (CASE) programs.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dore, E.M., Lyon, J.T. The Structures of Silicon Clusters Doped with Two Gold Atoms, Si n Au2 (n = 1–10). J Clust Sci 27, 1365–1381 (2016). https://doi.org/10.1007/s10876-016-1006-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-016-1006-y