
Abstract
Purpose
Adenosine deaminase 2 (ADA2) have been reported to cause vasculitic diseases and immunodeficiency recently. Patients present with stroke episodes and rashes mimicking polyarteritis nodosa (PAN). We report a patient who has been followed up with severe neutropenia and found an unexpectedly revealed novel mutation in CECR1 affecting ADA2.
Methods
We reviewed medical records and clinical history of the patient. No mutations in other known neutropenia genes such as ELA, G6PC3, HAX1, AP3B1, LAMTOR2, VPS13B, VPS45, GFI1, JAGN1, or WAS could be detected. Sanger sequencing was used to confirm the genetic variants in the patient and relatives.
Results
Genetic analysis by exome sequencing revealed a novel mutation in the gene CECR1 (c.G962A; p.G321E) which segregated perfectly in the relatives.
Conclusion
This is the first DADA2 patient presenting with severe neutropenia. We suggest that in patients with unexplained cytopenias combined with immunodeficiency, fevers of unknown origin and high inflammation markers, DADA2 should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CECR1 (cat eye syndrome chromosome region, candidate 1) mutations encoding adenosine deaminase 2 (ADA2) have been reported to cause vasculitic diseases with skin manifestations and immunodeficiency recently. Patients present with stroke episodes and rashes mimicking polyarteritis nodosa (PAN) [1, 2]. In many cases, the histological findings are consistent with the diagnosis of polyarteritis nodosa (PAN) with childhood-onset. Recent reports added more variable immunodeficiency phenotypes such as low or high Ig levels, in particular selective IgM deficiency, high IgG levels, pan-hypogammaglobulinemia-like CVID (common variable immunodeficiency), and low memory B cells. New clinical presentations such as red cell aplasia and pure antibody deficiency were also reported [3, 4]. By now, 19 different mutations in CECR1 gene have been detected. The pathogenetic mechanism of DADA2 is still unclear. Here, we would like to report a patient who has been followed up with severe neutropenia about 5 years. No mutations in other known neutropenia genes such as ELANE [5, 6], G6PC3 [7], HAX1 [8], AP3B1 [9], LAMTOR2 [10], VPS13B [11], VPS45 [12, 13], GFI1 [14], JAGN1 [15], or WASP [16] could be detected. Exome sequencing unexpectedly revealed a novel mutation in CECR1 affecting ADA2. Clinical findings of DADA2 were established in the last 1 year of the patient before she died.
Case Report
The patient was initially admitted to the hospital at the age of 7 years due to febrile neutropenia. Clinical history revealed frequent oral aphthous lesions and two skin abscesses in her early life. She also suffered from recurrent gastroenteritis. In the first laboratory examination, neutropenia was detected (absolute neutrophil count (ANC): 0–100/mm3). Severe neutropenia persisted in all blood counts (0–300/mm3), whereas other parameters were normal (Hb 12.1 g/dl, white blood cells (WBC) 7800/mm3, platelets (Plt) 349,000/mm3, absolute lymphocyte count 6500/mm3). Detailed immunological phenotyping uncovered no obvious alterations in lymphocyte subtypes, lymphocyte proliferation tests, neutrophil function tests, and immunoglobulin levels (Table 1, Fig. 1). Also peripheral blood smear and bone marrow aspiration did not show any alterations. Secondary causes (such as malignancy, myelodysplastic syndrome (MDS), severe infections) of neutropenia could be excluded. After initial hospitalization, she was consulting our clinics four to five times per year due to recurrent febrile neutropenia episodes. No other symptoms like abdominal pain, arthritis, or rashes except oral aphthous lesions appeared. Acute phase reactants were high in every attack but no culture growth was proved from any region of the body. We excluded malignancy, familial Mediterranean fever (FMF), and Behçet’s disease with some basal and genetic tests such as bone marrow aspiration, immunophenotyping of the bone marrow, imaging of the thorax, abdomen, and cranium, FMF mutation analysis, HLA tissue typing and pathergy test Mild abdominal pain accompanied with few febrile attacks was not remarkable. Nevertheless, colchicine was given empirically, but she did not respond. She was started on G-CSF prophylaxis which led to increase of ANC. Consequently, she received G-CSF treatment for 2 years.

Demonstration of absolute neutrophil counts follow
Despite treatment, she was required three to four times hospitalizations per year. At 9 years of age, she suffered from a life-threatening infection resulting in candida esophagitis and gastritis which ultimately led to fungal pneumonia. During this period, she further suffered from lymphopenia (ALC 200–1300/mm3). Myelodysplastic syndrome and malignancy were re-excluded. Although the laboratory and clinical findings did not suggest severe congenital neutropenia and cyclic neutropenia, we wanted to exclude this diagnosis by mutation analysis. All genes about congenital neutropenia were found to be normal.
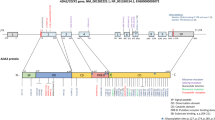
After this severe infection, genetic analysis was performed. Exome sequencing revealed a novel mutation in the gene CECR1 (c.G962A; p.G321E) which was segregating in the pedigree (Fig. 2). This mutation was predicted to be damaging with several prediction algorithms (Table 2). The patient was well about 1 year again until persistent fever of unknown origin and abdominal pain emerged. Based on the identification of ADA2 deficiency, we started anti-tumor necrosis factor (TNF)-α (etanercept) treatment paralleled by initiation of bone marrow transplant search. Only two doses of etanercept could be given; severe, watery diarrhea (40–50 times/day) started which caused transmission to the intensive care unit (ICU). Because of this, etanercept treatment was stopped. After 1 week in the ICU, a hemorrhagic necrosis appeared on her right lower leg and foot (Fig. 3). It progressed to the upper region of the leg and amputation was considered. Because of uncontrolled inflammation or infection, the patient deceased at the age of 11.

Chromatograms showing the mutation in CECR1

Hemorrhagic necrosis on right foot and leg
Discussion
Initially, deficiency of ADA2 (DADA2) was described in patients with vasculopathy and PAN-like skin manifestations. Patients with early-onset lacunar strokes have also been reported [1, 2]. In addition to variable reported immunodeficiencies, both IL17 receptor A and ADA2 mutations have also been described in the same patient [20]. In our patient, Ig levels were normal until the age of nine. At the age of 9 years, we noticed an increase in IgG levels and decrease in IgM levels. At the age of nine during her fatal hospitalization, general lymphopenia (500–1500/mm3) was detected without alterations of lymphocyte subtypes or proliferation capabilities. We excluded that the patient may suffer from myelodysplastic syndrome/acute myeloid leukemia (AML). Despite the fever of unknown origin and leukopenia, no autoantibodies were detected which ruled out the diagnosis of systemic lupus erythematosus. The encoded protein of ADA2 gene is one of the two adenosine deaminases found in humans, which regulate levels of the signaling molecule, adenosine. Secreted from monocytes undergoing differentiation and may regulate cell proliferation and differentiation, ADA2 is also responsible for catalyzing the conversion of adenosine to inosine. It binds to adenosine receptors on regulatory T cells expressing CD39 and helps immunological synapse formation. In a recent report, the authors found that ADA2 binds to neutrophils, monocytes, NK cells, and B cells [21]. A decrease in NK, NKT, and CD16 subset of monocytes was a characteristic feature of DADA2 patients in their study [21]. In our patient, all lymphocyte subset percentages stayed normal at different times of evaluation despite persisting lymphopenia.
ADA2 is expressed only in monocytes, macrophages, and dendritic cells, not in the endothelial cells [22]. This might explain why endothelial damage occurs in DADA2, but it remains still unclear why variable phenotypes are seen in different patients. In one report, CECR1 mutations were found to be associated with increased neutrophil counts in peripheral blood. Belot et al. proposed that ADA2 may act as a regulator of neutrophil activation, and that DADA2 results in endothelial damage via a neutrophil-driven process [23]. In another report, it was stated that ADA2 seemed to be involve in the balance between pro-inflammatory and anti-inflammatory monocytes; in the case of its absence, ADA2 was associated with a defect in differentiation of anti-inflammatory macrophages, which leads to a prevalence of pro-inflammatory cells [2, 24]. Montfrans et al. reported a DADA2 patient with granulocytopenia with accompanying hepatosplenomegaly and red cell aplasia [25]. Other cytopenias especially lymphopenia were reported in diagnosed DADA2 patients with vasculopathic manifestations [4, 7, 25,26,27,28]. Our patient initially presented with neutropenia as a different first manifestation. Other clinical findings consistent with DADA2 appeared about 4 years after the first hospitalization due to neutropenia. We might have speculated that the febrile neutropenia episodes could be caused by unidentified vasculopathic inflammation which is one of the hallmark features of DADA2. However, other symptoms like arthritis or stroke were not present in our patient. We want to emphasize that this was the first patient manifested with only neutropenia before other manifestations.
Our patient bears a novel homozygous mutation in the gene CECR1 (c.G962A; p.G321E). Although there is no established genotype-phenotype correlation, homozygous mutation R169Q in CECR1 seems more hematological manifestations than the other 18 types of mutations in a recent review of all reported patients [24]. We always thought that this patient had congenital neutropenia and hospitalizations due to unexplained infections. So, we can just speculate that the different first manifestation can be due to the location of the mutation. Further clinical studies are suggested in order to understand the phenotypic variability and the genotype-phenotype correlation [24].
In conclusion, our data and other reports [3, 4, 20, 24, 28] reveal an expanding spectrum of phenotypes in DADA2 in addition to the well-defined symptoms of vasculopathies, strokes, and/or PAN-like situations. As in our patient, neutropenia can be the first clinical manifestation of DADA2 which can be accompanied with other findings during follow-up. We suggest that in patients with unexplained isolated neutropenia/leucopenia combined with immunodeficiency, fevers of unknown origin, and high inflammation markers, DADA2 should be considered as a potential cause of the disease.
References
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31.
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20.
Hashem H, Egler R, Dalal J. Refractory pure red cell aplasia manifesting as deficiency of adenosine deaminase 2. J Pediatr Hematol Oncol. 2017;39(5):e293–6.
Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. 2016;36(3):179–86.
Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110(13):4179–87.
Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, Ballmaier M. The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum Mutat. 2013;34(6):905–14.
Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360(1):32–43.
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schäffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia [Kostmann disease]. Nat Genet. 2007;39(1):86–92.
Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3(1):11–21.
Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schäffer AA. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13(1):38–45.
Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Träskelin AL, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72(6):1359–69.
Stepensky P, Saada A, Cowan M, Tabib A, Fischer U, Berkun Y, et al. The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy. Blood. 2013;121(25):5078–87.
Vilboux T, Lev A, Malicdan MC, Simon AJ, Järvinen P, Racek T, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N Engl J Med. 2013;369(1):54–65.
Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34(3):308–12.
Boztug K, Järvinen PM, Salzer E, Racek T, Mönch S, Garncarz W, et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat Genet. 2014;46(9):1021–7.
Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27(3):313–7.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome aggregation, analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Fellmann F, Angelini F, Wassenberg J, Perreau M, Arenas Ramirez N, Simon G, et al. IL-17 receptor A and adenosine deaminase 2 deficiency in siblings with recurrent infections and chronic inflammation. J Allergy Clin Immunol. 2016;137(4):1189–1196.e2.
Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. 2017;74(3):555–70.
Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. 2010;88(2):279–90.
Belot A, Wassmer E, Twilt M, Lega JC, Zeef LA, Oojageer A, et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatr Rheumatol Online J. 2014;12:44.
Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. 2016;14(1):51.
van Montfrans J, Zavialov A, Zhou Q. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):478.
Van Eyck L Jr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. 2015;135(1):283–7.e5.
Kastner DL, Zhou Q, Aksentijevich I. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):480–1.
Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of adenosine deaminase type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheumatol. 2016;68(9):2314–22.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Cipe, F.E., Aydogmus, C., Serwas, N.K. et al. Novel Mutation in CECR1 Leads to Deficiency of ADA2 with Associated Neutropenia. J Clin Immunol 38, 273–277 (2018). https://doi.org/10.1007/s10875-018-0487-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-018-0487-x