
Abstract
Background
Pulmonary disease is common in patients with common variable immunodeficiency disorders (CVID) and involves infections, chronic airway disease and interstitial lung disease. Chronic pulmonary disease is associated with excess morbidity and early mortality and therefore early detection and monitoring of progression is essential.
Methods and Purpose
Thin slice CT scan and pulmonary function were used to determine the prevalence and spectrum of chronic (pre-clinical) pulmonary disease in adult CVID patients regardless of symptoms. CT Scans were scored for airway abnormalities (AD) and interstitial lung disease (ILD). Other CVID related complications and B and T lymphocyte subsets were analyzed to identify patients at risk for pulmonary disease.
Results
Significant pulmonary abnormalities were detected in 24 of the 47 patients (51 %) consisting of AD in 30 % and ILD in 34 % of cases. In only 7 (29 %) of these 24 patients pulmonary function test proved abnormal. The presence of AD was correlated to (recurrent) lower respiratory tract infections despite IgG therapy. The presence of ILD was correlated to autoimmune disease and a reduction in the numbers of CD4 + T cells, naïve CD4 + T cells, naïve CD8 + T cells and memory B cells and lower IgG through levels over time.
Conclusion
Preclinical signs of AD and ILD are common in CVID patients despite Ig therapy and do not correlate to pulmonary function testing. Patients at risk for ILD might be identified by the presence of autoimmunity or a deranged T cell pattern. Larger studies are needed to confirm these findings and to determine thresholds for the T lymphocyte subsets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Common Variable Immunodeficiency disorders (CVID) is a descriptive term for a heterogeneous group of patients with a primary antibody deficiency syndrome with the common denominator of recurrent respiratory tract infections and hypogammaglobulinaemia caused by B cell dysfunction [1]. In addition, T cell abnormalities have been demonstrated in some patients. Recently patients with late onset combined immunodeficiency (LOCID) were described as a distinct group of patients who differ from classic CVID through clinical presentation and immunological phenotype [2]. Complications due to immune dysregulation are common in CVID patients and involve lymphoproliferative disease, autoimmunity, enteropathy and malignancies [3, 4]. Chronic pulmonary disease is most frequent in CVID involving asthma and chronic obstructive pulmonary disease [5], structural airway disease (AD) and interstitial lung disease (ILD) [5]. Structural airway disease is generally the result of recurrent lower respiratory tract infections leading to airway wall thickening, air trapping and bronchiectasis. Bronchiectasis has been described in 4–76 % of CVID patients [3, 6–11] and has been associated with recurrent infections, a delayed diagnosis of CVID and inadequate treatment [7, 8]. Immunoglobulin replacement therapy and prophylactic antibiotics have proven to be effective in preventing pulmonary infections [12–14] and may thus indirectly prevent (the worsening of) structural airway disease (e.g. bronchiectasis) [15–17]. However, the development of structural airway disease seems to progress despite adequate immunoglobulin replacement and even in the absence of clinically recognized infections [7, 9], suggesting that additional factors contribute to its development which need further clarification. Interstitial lung disease (ILD) includes granulomatous lung disease, lymphoid interstitial pneumonia, organizing pneumonia and lymphoproliverative disorders. ILD is characterized by nodules, ground glass phenomena, and reticulations on CT scans [18] and may be caused by immune dysregulation associated with B - and T-lymphocyte dysfunction and consequent dysregulation of cytokines (interleukin-6 and TNFα) [19–21]. Steroids seems to be effective in CVID patients for the treatment of ILD and cyclosporine and monoclonal antibodies against TNFα may be considered as alternatives, although studies are lacking [22–24]. In a retrospective analysis of seven CVID children with ILD it was found that combination chemotherapy Rituximab with azathioprine resulted in a significant improvement of pulmonary function and parenchymal abnormalities found on HRCT [25]. However additional studies will be necessary to assess the long-term effect.
Chronic pulmonary disease in CVID patients has been associated with an increased mortality [3] and therefore early detection and monitoring of progression will be essential to prevent progressive lung disease by additional therapeutic measures. Earlier studies support the use of an initial chest computed tomography (CT) at the time of CVID diagnosis and recommendations for follow up CT scans range between 1–5 years to evaluate the development or progression of pulmonary disease [7, 26]. Alternatively, to minimize the risks of X-ray exposure in CVID patients [27] CT scanning at intervals of 4–5 years with interim annual pulmonary function testing may be more appropriate [9, 28, 29]. However, the correlation between pulmonary function testing and findings on CT is often weak [30, 31]. To minimize the risk of X-ray exposure, it is desirable to select patients at risk for chronic pulmonary disease based on clinical or immunological parameters.
The purpose of this study was to describe the prevalence of preclinical pulmonary abnormalities in CVID patients by thin slice CT and a standardized CT scan scoring system recently described in paediatric CVID patients [32, 33] and to correlate these findings with the results of pulmonary function tests, IgG trough levels, clinical parameters and peripheral blood B- and T lymphocyte subsets.
Methods and Materials
Study Population
Between 2008 and 2012 47 of the 61 adult CVID patients in care at the department of Internal Medicine and Infectious diseases of the University Medical Centre Utrecht were screened for pulmonary disease as part of a structured follow-up by thin-slice computed tomography (CT) scan of the chest and pulmonary function testing. The CT scans were performed during routine follow-up at the outpatient clinic (screening). We did not use questionnaires to assess complaints. The remaining 14 patients had recently undergone conventional CT scanning or refused for various reasons. All patients had been diagnosed according to the European Society for immune deficiencies (ESID)/Pan-American Group criteria for Immunodeficiency (PAGID) [1, 34]. Recently patients with late onset combined immunodeficiency (LOCID) were described [2], however no single patient in our cohort fitted the criteria for a LOCID. Explicit approval of the Ethic committee was not necessary since monitoring of immunoglobulin titers, and performing of T- and B cell phenotyping and CT scanning are considered standard care for CVID patients [9, 28, 35, 36].
Clinical Evaluation
The medical hospital records of all patients were evaluated for clinical and laboratory data and the results of prior imaging. The time to CVID diagnosis was defined as the time (yrs.) between the year of onset of disease-related symptoms and the year of diagnosis. A pneumonia was defined according to the SWAB/NVALT [37] or alternatively as a physician-diagnosed pneumonia. Chronic obstructive pulmonary disease (COPD) was defined according to the GOLD criteria [38]. Asthma was defined according to the Ginasthma guidelines [39]. Interstitial pulmonary disease was classified as granulomatous lung disease, lymphoid interstitial pneumonia, organizing pneumonia or lymphoproliverative disorder of the lung. granulomatous lung disease was confirmed with biopsy whereas the other conditions were radiological diagnoses.[40] Other CVID-related complications were classified according to earlier reports by Chapel [3, 4], I. autoimmunity (e.g. auto-immune cytopenia and organ- specific autoimmunity); II. polyclonal lymphocytic infiltration, or granulomatous disease or hepatosplenomegaly III. enteropathy; IV. malignancies.
Laboratory Data
In all patients IgG trough levels had been measured once or twice a year. The median IgG trough level over a given set of years was defined as the average IgG level per year divided by the number of years. T- and B cell phenotyping was performed in patients between 2007 and 2011 in clinically stable patients. The mean time since diagnosis of the patients at that time was 11,7 years (2–30 years). Therefore, in some patients phenotyping was performed several years after diagnosis and in some patients at the diagnosis.
The T- and B cell (sub) populations were analyzed by four-color flow cytometry using whole blood and antibodies to CD3, CD45, CD27, CD4, CD8, HLA-DR, CD38, CD45RA and CD19, CD27, CD38, CD10, IgM, IgG, IgA and IgD, respectively, as described previously [41]. Within the CD19 + B cell compartment, the following populations were distinguished: IgD + CD10 + CD38++ recent bone-marrow emigrants (RBE), IgM + IgD + CD27-CD10- naive B cells, non-Ig class-switched IgM + IgD + CD27+ memory B cells, IgG + CD27+ and IgA + CD27+ memory B cells. CD3+ T cells were divided into CD4+ and CD8+ subsets, and subsequently into CD38 + HLA-DR + activated T cells, CD45RA + CD27+ naïve, and non-naïve CD45RA + CD27-, CD45RA-CD27+ or CD45RA-CD27- T cells. IgG trough levels were measured once or twice a year in clinically stable patients during follow up, and more frequent in patients who were diagnosed with disease-related complications. In our current daily practice we aim to reach IgG through levels of at least 8 g/L [12].
Thin Slice CT and Scoring
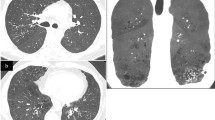
Thin slice CT followed a routine protocol using a 16-detector-row CT scanner (MX8000 IDT or Brilliance-16), a 64-detector-row CT scanner (Brilliance-64) or a 256-detector-row scanner (Brilliance iCT, all Philips Healthcare, Best, The Netherlands). Inspiratory scans were acquired in a caudocranial direction using 130 mAs and 100–120 kVp (depending on body weight) and expiratory scans in a caudocranial direction using 20 mAs and 80–100 kVp (depending on body weight). Images were reconstructed with a slice thickness of 1-mm (16-detector-row and 256-detector-row scanners) or 0.9-mm (64-detector-row scanner) and an increment of 0.7-mm. The presence of structural airway disease (AD) and interstitial lung disease (ILD) was scored in each lobe by an independent observer (P. de J.) according to a previous published scoring system [32, 33, 42]. Signs of AD (the CVID airway disease score ) were scored by assessing the presence of bronchiectasis, airway wall thickening, mucus plugging, tree-in-bud and air-trapping. Signs of ILD (the CVID interstitial disease score) were scored by assessing the presence of opacities, ground glass, septa thickening and lung nodules. As previously described an ILD score >5 and an AD score >7 was defined as clinically significant [32, 33].
Pulmonary Function Testing
All pulmonary function tests were performed using standardized equipment according to the current European Respiratory Society (ERS)/American Thoracic society guidelines [43–45] and consisted of spirometry and body plethysmography (forced expiratory volume in one second (FEV1), forced vital capacity (FVC), FEV1/FVC ratio, vital capacity (VC), residual volume (RV), total lung capacity (TLC) and transfer coefficient for carbon monoxide (Kco). All Spiro metric values were obtained after inhalation of a bronchodilator. All values are presented as percentages of the predicted values [46]. The lower limits of normal were calculated using the reference equations of the European Community of Coal and Steel (ECCS) [46]. Airway obstruction was diagnosed when the FEV1/FVC was <80 % of the predicted value. Diffusion capacity was considered abnormal when measured <75 % of the predicted value [46, 47]. Each test was evaluated by an independent observer (J.W.L).
Data Analysis
Data were analyzed using SPSS 20.0 (SPSS Inc.; Chicago, IL). Continuous, non-normally distributed variable were analyzed by using the Mann–Whitney U tests. Chi square tests were performed for categorical variables. Statistical significance was determined at p < 0.05. An analysis of the sensitivity, specificity and positive predictive value of the specific immunological markers for ILD was performed. For this analysis we used an ILD score >5 as golden standard. As the choice of a single cut-off value is arbitrary, we also calculated the c-statistic to give an overall measure of the discriminative power of the variables.
Results
Pulmonary Pathology Detected by Thin Slice CT
Between 2008 and 2012 47 of the 61 patients in care were evaluated by thin slice CT scan (6 patients without expiration and 41 patients with expiration). The baseline characteristics of the patients are shown in Table I. The median age was 37 years (IQR 29–54 years.) at the time of the CT scan. The median time to CVID diagnosis had been 9.5 years (IQR 5–16 years.). Half of the patients (23/47, 49 %) had been diagnosed with one or more CVID related complications such as auto immune- or lymphoproliverative conditions. Sixteen of the 47 participating patients (34 %) had already been diagnosed with a symptomatic pulmonary condition of which nine (19 %) with asthma, three with ILD (6 %) compared to 43 % of all CVID patients in care. Three patients (6 %) had already been diagnosed with bronchiectasis. One of the patients died from a non-pulmonary cause. In 10 of the 47 patients a conventional CT scan was available ranging between 3.5–14.5 years ago, however, the CT scoring system from the current study could not be applied to earlier conventional CT scans and therefore progress could not be assessed.
In 45 of 47 CVID (96 %) patients one or more CT abnormalities were detected (Table II). In 40 of 47 (85 %) patients the structural airway disease (AD) score was ≥1, which was most commonly based on air wall thickening (35 patients, 75 %) and/or the presence of bronchiectasis. In 30 patients (64 %) signs of bronchiectasis were found. In 34 patients (72 %) any signs of ILD score was detected (score ≥1), which was most commonly based on the presence of nodules (26/47patients, 55 %).
An AD score >7 or an ILD score >5 was considered indicative of significant pathology based on an earlier report using this scoring system [32, 33]. In 24 patients (51 %) CT abnormalities were considered significant for AD, ILD or both. In 14 of the 47 patients (30 %) the AD score was significant of which four patients had already been diagnosed with bronchiectasis. The ILD score was significant in 16 of 47 patients (34 %) of which three patients had been diagnosed earlier with ILD.
Pulmonary Pathology in Relation to Clinical Characteristics
We did not find a relation between the stage of disease and the median time (yrs.) since start of the actual disease nor the median time (yrs.) since diagnosis of CVID.
Tables III and IV show the clinical differences between patients with and without significant AD or ILD, respectively. No correlation was found between current age, the age at diagnosis or the time to diagnosis on the one hand and the presence of significant AD or/ILD abnormalities (AD score >7; ILD score >5) on the other hand. Patients with an AD score >7 had suffered more frequently from (recurrent) pneumonia when compared to patients with an AD score ≤ 7 (p = 0.009). Notably, bronchiectasis was also detected in patients who had never been diagnosed with pneumonia (17 of the 30 patients with bronchiectasis, 57 %).
In patients with an ILD score >5 autoimmune disease was more frequent when compared to patients with an ILD score ≤ 5 (6/16 (38 %) vs. 2/31 (6 %) patients, p = 0.007). In the group of patients without any CVID-related complications the majority had an ILD score <5 (21 (68 %) vs. 3 patients with an ILD score >5 (19 %, p = 0.001).”
Pulmonary function testing proved abnormal in 10 of the 44 tested patients (Tables II, III and IV). In only 7 (29 %) of the 24 patients with significant CT abnormalities pulmonary function testing proved abnormal indicating the poor correlation of pulmonary function testing and findings on the CT scan.
Pulmonary Pathology in Relation to Immunological Characteristics
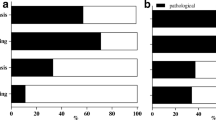
The distribution of IgG levels at the time of CVID diagnosis and the median IgG trough levels during therapy in the last ten years did not differ between patients with or without any significant AD or ILD. However we found that the presence of ILD was correlated to a prolonged time (>1 year) of IgG trough levels <8 g/L (Fig. 1).

Correlation between the presence of ILD and the IgG trough level. This figure shows de percentage of patients with and without prolonged time of inadequate IgG trough levels, with and without significant ILD (ILD score >5). The median IgG trough level over a given set of years was defined as the average IgG level per year divided by the number of years. ILD: interstitial lung disease
In patients with any AD the majority (67 %) had IgG trough levels that were considered adequate (>8 g/L).
Patients with an ILD score >5 showed significantly different T- and B cell profiles when compared to patients with an ILD score ≤ 5 (Table V and Fig. 2). The median absolute number of CD4+ T cells was lower in patients with an ILD score >5 when compared to patients with an ILD score ≤ 5 (601/mm3 vs. 989 mm3; p = 0.03). This reduction was mainly related to lower absolute numbers of naïve CD4+ T cells (78 vs. 434 cells/mm3; p = 0.004) and effector/memory CD4 + T cells (383/mm3; vs. 529/mm3; p = 0.01). Additionally, the absolute numbers of naïve CD8 + T cells were reduced in the group of patients with significant signs of ILD when compared to the patients without (Table V). Patients with an ILD score >5 also showed lower numbers of naïve B cells and IgG memory B cells when compared to patients with an ILD score ≤ 5 (naïve B cells: 78/mm3 vs. 162/mm3; p = 0.03 and IgG memory B cells: 1.5/mm3 vs. 7/mm3; p = 0.04). Furthermore, the absolute number of class switched memory B cells were reduced in the patients with an ILD score >5 when compared to the patients with a score ≤ 5 (6/mm3 vs. 11/mm3 respectively; p = 0.03). The absolute number of CD4+ T cells and the naïve CD4 + T cells were found to have the highest sensitivity (80 % and 82 %, respectively) and positive predictive values (60 % and 64 % respectively) to identify patients at risk for ILD.

Median count of lymphocytes within the T- and B cell subset in patients with and without ILD. Boxplot: boxes indicates 50th percentile, whispers indicate the 25th and 75th percentile. CVID: Common variable immunodeficiency disorders; ILD: interstitial lung disease, ILD score >5 *Median absolute counts per mm3. Reference values, (all in absolute numbers): From: van Gent et al., Clinical Immunology (2009). CD3+ T lymphocytes (100–400); CD4 T + lymphocytes (400–1,300); Naive (CD45RA + CD27+) CD4+ T lymphocytes (240–790); Effector/memory CD4+ T lymphocytes (150–500); Naive (CD45RA + CD27+ ) CD8 + T lymphocytes (220–400); Naief (IgM + IgD + CD27-CD10-) B cells (72–257); IgG (IgG + CD27+) memory B cells (2–51); non-Ig class-switched IgM + IgD + CD27+ memory B cells, IgG + CD27+ and IgA + CD27+ memory B cells
Patients with or without significant AD abnormalities (Table VI) and patients with and without bronchiectasis did not differ in T- and B cell numbers.
Discussion
Chronic pulmonary disease is a common complication in CVID patients and may develop despite adequate immunoglobulin substitution therapy [7–9, 11, 48]. Given the associated morbidity and mortality [3], early detection and monitoring seems essential.
This cross-sectional study shows a high incidence of (pre-clinical) pulmonary CT abnormalities in patients with CVID despite adequate immunoglobulin substitution therapy. Subtle pre-clinical pulmonary abnormalities were detected in virtually all participating CVID patients by thin slice CT scan (AD or ILD scores ≥ 1 in 96 % and 51 % of patients, respectively). However, significant abnormalities (AD score >7) or ILD (ILD score >5) were present in 30 % and 34 % of patients, respectively. These rates might be an underestimation since the percentage of patients with established pulmonary disease that participated in the study was lower (34 %) when compared to the entire group of 61 CVID patients in care (43 %). When compared to children these rates are considerably higher in adult CVID patients. A recent publication that used the same scoring system in CVID children in our center reported significant airway abnormalities in 20 % and ILD in 14 % of cases [32, 33].
Bronchiectasis and structural airway disease have been associated with (recurrent) lower respiratory tract infections [49, 50] and immunoglobulin replacement therapy (with adequate IgG trough levels) and prophylactic antibiotics have proven to be effective in preventing pulmonary infections [12–14]. In our study the presence of significant AD (AD score >7) was indeed associated with the occurrence of pneumonia after initiation of IgG therapy. However, in our analysis 17 of the 30 patients with bronchiectasis had never been diagnosed with pneumonia, indicating that infections might have occurred subclinical. Three prior publications also demonstrated (silent) progression of structural airway disease despite adequate therapy and in the absence of pulmonary infections [9, 15, 51]. It has been proposed that IgG trough levels >9 g/L should be attained in patients with bronchiectasis, however, evidence supporting this proposition is scarce [15, 52]. In earlier studies bronchiectasis was shown to be related to a reduced number of B memory cells [53–55], which could not be confirmed in our study, nor a relation to T cell disorders.
It therefore seems that AD develops mainly due to the cumulative effect of infections.
Concurrent with earlier reports, ILD was associated with the presence of auto immune disease and enteropathy in our study. Reversely, of the patients with an ILD score ≤5 the majority had no CVID related complications. Furthermore, CVID patients with significant signs of ILD were found to have a distinct cellular distribution of T- and B cell subpopulations when compared to patients without ILD. In ILD patients the absolute numbers of CD4 + T cells, naive CD4 + T cells, and naïve and switched memory B cells were markedly lower. Earlier reports that describes the classification of the CVID population according to B cell phenotype [56–58] have shown that patients with more severe complications in general had lower numbers of switched memory B cells [53, 56, 58–60] which was associated with granulomatous-, autoimmune disease and splenomegaly [58, 61]. Moreover, a reduction of CD4 naive T cells has been most consistently associated with CVID related complications such as autoimmunity and lymphoproliferation [60, 62], splenomegaly and granulomatous disease [63]. Only a few studies have explored T cell phenotype in adult CVID patients with pulmonary disease. An earlier study showed a different T cell pattern in children with ILD when compared to our study in adults. In that particular study, the differences between children with and without ILD were detected in the CD8+ T cell compartment only [33]. Evidently, differences in the T cell distribution exist between age-groups, since the memory subset will gradually increase during aging. Furthermore, in general, children will experience more viral infections and therefore an increased activation pattern can be expected in the CD8 + T cell compartment. The decreased thymic output and decreased number of regulatory T cells in CVID patients [63, 64] could account for the auto inflammatory conditions, including ILD. In children, a lower number of memory B cells has also been associated to ILD [33] and other CVID related complications [56, 58, 65]. The lack of B cells might contribute to ILD by the increased prevalence of infections, which may evoke exacerbations of auto-inflammatory disease.[66]. However, ILD occurs in varied clinical settings, including infection and immune system dysfunction, it is thought to represent a nonspecific response to multiple stimuli [67].
Interestingly, we found that patients with IgG trough levels <8 g/L for more than one year were more likely to have significant ILD-related abnormalities. It is known that immunoglobulin therapy has also shown to be beneficial in autoimmune disease [68]. However, the doses used in those treatments are usually much higher than those used for replacement therapy and the mechanism of its immunomodulatory and anti-inflammatory effects remains unclear [68]. It is however known, that recurrent infections - which are clearly related to insufficient IgG trough levels in CVID- can evoke exacerbations of inflammatory diseases [66].
Early detection and monitoring of chronic pulmonary disease in CVID patients seems essential. Our study indicates that pulmonary function testing is a weak predictor of early pulmonary abnormalities. Therefore, repetitive CT scanning and CT scoring systems could be reliable tools to monitor progression of pulmonary abnormalities and to identify patients at risk. However, no consensus has yet been reached about the frequency of CT scanning. In the current practice the current time interval for CT evaluation ranges from 1 to 5 years [7, 26]. Based on our and others research we would suggest biannual CT screening of CVID patients with (I) active autoimmune disease or other non-infectious inflammatory disease, (II) patients with on-going frequent lower respiratory tract infections and (III) patients with a significant AD (>7) or ILD score (>5). For other CVID patients CT screening once per five years might be sufficient. The absolute number of the total CD4+ T cells count and the naïve CD4+ T cells and switched memory B cells might also be candidate immunological markers to identify patients at risk for ILD. However, a clear threshold (cutoff) for the immune-phenotypic markers could not be identified in our study.
Our study has the following limitations. First, the number of patients was too small to develop a reliable prediction model to define risk factors for pulmonary complications and therefore our findings require confirmation in larger studies. Second, it is a cross-sectional study and a prospective follow-up study will be necessary to establish progression of preclinical pulmonary abnormalities, the development of clinical disease and the relation to risk factors. Third, due to the low number of patients with prior thin slice CT scanning we could not establish at what point in time the CT abnormalities had evolved (before or after the initiation of immunoglobulin therapy) nor could progression be evaluated.
In conclusion, our study shows a relatively high frequency of preclinical pulmonary CT abnormalities in CVID patients despite IgG therapy and normal pulmonary function testing which emphasizes the need for screening with CT in this group of patients. Structural irway disease is correlated to pulmonary infections, but can also develop sub clinically. ILD is correlated to the presence of autoimmune disease and have a distinct cellular distribution of T and B cell subpopulations and lower IgG through levels over time. However, larger and prospective studies are required to determine cut off values for T and B cell markers to discern patients at risk for ILD and to ascertain the optimal follow up frequency aimed at preventing morbidity and mortality in CVID patients.
References
Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
Malphettes M, Gerard L, Carmagnat M, Mouillot G, Vince N, Boutboul D, et al. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis. 2009;49(9):1329–38.
Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86.
Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol 2012 Jul 19
Hampson FA, Chandra A, Screaton NJ, Condliffe A, Kumararatne DS, Exley AR, et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol. 2012;67(6):587–95.
Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92(1):34–48.
Kainulainen L, Varpula M, Liippo K, Svedstrom E, Nikoskelainen J, Ruuskanen O. Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol. 1999;104(5):1031–6.
Martinez Garcia MA, De R, Nauffal M, Munoz Pamplona MP, Compte TL, Macian V, et al. Respiratory disorders in common variable immunodeficiency. Respir Med. 2001;95(3):191–5.
Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27(3):308–16.
Oksenhendler E, Gerard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46(10):1547–54.
Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM. 2002;95(10):655–62.
Eijkhout HW, van der Meer JW, Kallenberg CG, Weening RS, van Dissel JT, Sanders LA, et al. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. a randomized, double-blind, multicenter crossover trial. Ann Intern Med. 2001;135(3):165–74.
Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109(6):1001–4.
Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the primary immunodeficiency committee of the American academy of allergy, asthma and Immunology. J Allergy Clin Immunol. 2006;117(4 Suppl):S525–53.
Davies G, Wilson R. Prophylactic antibiotic treatment of bronchiectasis with azithromycin. Thorax. 2004;59(6):540–1.
De GJ, Vendrell M, Alvarez A, Pallisa E, Rodrigo MJ, la RD D, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol. 2004;4(6):745–53.
Maarschalk-Ellerbroek LJ, Hoepelman IM, Ellerbroek PM. Immunoglobulin treatment in primary antibody deficiency. Int J Antimicrob Agents. 2011;37(5):396–404.
Touw CM, Van DV, De Jong PA, Terheggen-Lagro S, Beek E, Sanders EA, et al. Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy Immunol. 2010;21(5):793–805.
Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997;159(12):6236–41.
Mullighan CG, Marshall SE, Bunce M, Welsh KI. Variation in immunoregulatory genes determines the clinical phenotype of common variable immunodeficiency. Genes Immun. 1999;1(2):137–48.
Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127(8 Pt 1):613–7.
Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax. 2000;55(1):88–90.
Smith KJ, Skelton H. Common variable immunodeficiency treated with a recombinant human IgG, tumour necrosis factor-alpha receptor fusion protein. Br J Dermatol. 2001;144(3):597–600.
Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol. 2010;134(2):97–103.
Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol. 2013;33(1):30–9.
Deane S, Selmi C, Naguwa SM, Teuber SS, Gershwin ME. Common variable immunodeficiency: etiological and treatment issues. Int Arch Allergy Immunol. 2009;150(4):311–24.
Vorechovsky I, Scott D, Haeney MR, Webster DA. Chromosomal radiosensitivity in common variable immune deficiency. Mutat Res. 1993;290(2):255–64.
Cunningham-Rundles C. How I, treat common variable immune deficiency. Blood. 2010;116(1):7–15.
Yong PF, Tarzi M, Chua I, Grimbacher B, Chee R. Common variable immunodeficiency: an update on etiology and management. Immunol Allergy Clin North Am. 2008;28(2):367. x.
Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114(2):415–21.
Watts WJ, Watts MB, Dai W, Cassidy JT, Grum CM, Weg JG. Respiratory dysfunction in patients with common variable hypogammaglobulinemia. Am Rev Respir Dis. 1986;134(4):699–703.
Van DV, Van Montfrans JM, Terheggen-Lagro SW, Beek FJ, van Konijnenburg DP H, Kessels OA, et al. A CT scan score for the assessment of lung disease in children with common variable immunodeficiency disorders. Chest. 2010;138(2):371–9.
Van DV, de Jong PA, van Konijnenburg DP H, Kessels OA, Boes M, Sanders EA, et al. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol. 2011;165(2):235–42.
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing pagid (pan-american group for immunodeficiency) and esid (european society for immunodeficiencies). Clin Immunol. 1999;93(3):190–7.
Salzer U, Warnatz K, Peter HH. Common variable immunodeficiency - an update. Arthritis Res Ther. 2012;14(5):223.
Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107.
Wiersinga WJ, Bonten MJ, Boersma WG, Jonkers RE, Aleva RM, Kullberg BJ, et al. SWAB/NVALT (dutch working party on antibiotic policy and dutch association of chest physicians) guidelines on the management of community-acquired pneumonia in adults. Neth J Med. 2012;70(2):90–101.
Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease, GOLD Executive Summary. Am J Respir Crit Care Med 2012 Aug 9
http://www.ginasthma.org/. 2013. Ref Type: Generic
Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122(6):2150–64.
Van GR, van Tilburg CM, Nibbelke EE, Otto SA, Gaiser JF, Janssens-Korpela PL, et al. Refined characterization and reference values of the pediatric T- and B-cell compartments. Clin Immunol. 2009;133(1):95–107.
Brody AS, Kosorok MR, Li Z, Broderick LS, Foster JL, Laxova A, et al. Reproducibility of a scoring system for computed tomography scanning in cystic fibrosis. J Thorac Imaging. 2006;21(1):14–21.
Miller MR, Crapo R, Hankinson J, Brusasco V, Burgos F, Casaburi R, et al. General considerations for lung function testing. Eur Respir J. 2005;26(1):153–61.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38.
MacIntyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26(4):720–35.
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. report working party standardization of lung function tests, european community for steel and coal. official statement of the european respiratory society. Eur Respir J Suppl. 1993;16:5–40.
Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–55.
Gregersen S, Aalokken TM, Mynarek G, Kongerud J, Aukrust P, Froland SS, et al. High resolution computed tomography and pulmonary function in common variable immunodeficiency. Respir Med. 2009;103(6):873–80.
King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW. Characterisation of the onset and presenting clinical features of adult bronchiectasis. Respir Med. 2006;100(12):2183–9.
King P, Holdsworth S, Freezer N, Holmes P. Bronchiectasis. Intern Med J. 2006;36(11):729–37.
Gregersen S, Aalokken TM, Mynarek G, Fevang B, Holm AM, Ueland T, et al. Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol. 2010;104(6):503–10.
Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354–60.
Alachkar H, Taubenheim N, Haeney MR, Durandy A, Arkwright PD. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency. Clin Immunol. 2006;120(3):310–8.
Detkova D, De GJ, Lopes-da-Silva S, Vendrell M, Alvarez A, Guarner L, et al. Common variable immunodeficiency: association between memory B cells and lung diseases. Chest. 2007;131(6):1883–9.
Aydogan M, Eifan AO, Gocmen I, Ozdemir C, Bahceciler NN, Barlan IB. Clinical and immunologic features of pediatric patients with common variable immunodeficiency and respiratory complications. J Investig Allergol Clin Immunol. 2008;18(4):260–5.
Piqueras B, Lavenu-Bombled C, Galicier L, der Bergeron-van CF, Mouthon L, Chevret S, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23(5):385–400.
Ferry BL, Jones J, Bateman EA, Woodham N, Warnatz K, Schlesier M, et al. Measurement of peripheral B cell subpopulations in common variable immunodeficiency (CVID) using a whole blood method. Clin Exp Immunol. 2005;140(3):532–9.
Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85.
Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128(3):314–21.
Mouillot G, Carmagnat M, Gerard L, Garnier JL, Fieschi C, Vince N, et al. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. 2010;30(5):746–55.
Berglund LJ, Wong SW, Fulcher DA. B-cell maturation defects in common variable immunodeficiency and association with clinical features. Pathology. 2008;40(3):288–94.
Bateman EA, Ayers L, Sadler R, Lucas M, Roberts C, Woods A, et al. T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin Exp Immunol. 2012;170(2):202–11.
Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178(6):3932–43.
Melo KM, Carvalho KI, Bruno FR, Ndhlovu LC, Ballan WM, Nixon DF, et al. A decreased frequency of regulatory T cells in patients with common variable immunodeficiency. PLoS One. 2009;4(7):e6269.
Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27 (+) IgM (−) IgD (−)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99(5):1544–51.
Sener AG, Afsar I. Infection and autoimmune disease. Rheumatol Int. 2012;32(11):3331–8.
Fishback N, Koss M. Update on lymphoid interstitial pneumonitis. Curr Opin Pulm Med. 1996;2(5):429–33.
Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367(21):2015–25.
Grant Support
L.J. Maarschalk-Ellerbroek was supported by a non-restricted educational grant from Baxter Bioscience.
Author Contributions
L.J. Maarschalk-Ellerbroek: study concept and design, administrative acquisition of data, data entry, analysis and interpretation of data, statistical analysis, drafting of the manuscript and final approval of the version to be published.
P.A.de Jong: acquisition of data (evaluation and analyzing all CT scans results), critical revision of the manuscript for important intellectual content and final approval of the version to be published.
J. Montfrans: critical revision of the manuscript for important intellectual content and final approval of the version to be published.
J.W. Lammers: acquisition of data (evaluation and analyzing all pulmonary function tests results), critical revision of the manuscript for important intellectual content and final approval of the version to be published.
A.C. Bloem: acquisition of data (evaluation and analyzing all B- and T phenotyping results), critical revision of the manuscript for important intellectual content and final approval of the version to be published.
A.I.M. Hoepelman: study supervision, critical revision of the manuscript for important intellectual content and final approval of the version to be published.
P.M. Ellerbroek: study concept and design, study supervision, analysis and interpretation of data, statistical analysis, and drafting of the manuscript, critical revision of the manuscript for important intellectual content and final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maarschalk-Ellerbroek, L.J., de Jong, P.A., van Montfrans, J.M. et al. CT Screening for Pulmonary Pathology in Common Variable Immunodeficiency Disorders and the Correlation with Clinical and Immunological Parameters. J Clin Immunol 34, 642–654 (2014). https://doi.org/10.1007/s10875-014-0068-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-014-0068-6