
Abstract
Purpose
This study reports the identification of a novel heterozygous IKBA missense mutation (p.M37K) in a boy presenting with ectodermal dysplasia with immunodeficiency (EDA-ID) who had wild type IKBKG gene encoding NEMO. Our aim was to characterize the clinical course of this IκB-α gain-of-function mutant and to investigate if the p.M37K substitution affects NF-κB activation by interfering with IκB-α degradation, thus impairing NF-κB signaling and causing the EDA-ID phenotype.
Methods
NF-κB signaling was evaluated by measuring IκB-α degradation in patient fibroblasts. In addition, transiently transfected HeLa cells expressing either the M37K-mutant IκB-α allele, the previously characterized S36A-mutant IκB-α allele, or wild type IκB-α were evaluated for IκB-α degradation and NF-κB nuclear translocation following stimulation with TNF-α.
Results
Clinical findings revealed a classical ectodermal dysplasia phenotype complicated by recurrent mucocutaneous candidiasis, hypothyroidism, hypopituitarism, and profound combined immunodeficiency with decreased numbers of IL-17 T cells. IκB-α degradation after TNF-α and TLR agonist stimulation was abolished in patient fibroblasts as well as in HeLa cells expressing M37K-IκB-α similar to cells expressing S36A-IκB-α resulting in impaired nuclear translocation of NF-κB and reduced NF-κB dependent luciferase activity compared to cells expressing wild type IκB-α. Patient whole blood cells failed to secrete IL-6 in response to IL-1ß, Pam2CSK4, showed reduced responses to LPS and PMA/Ionomycin, and lacked IL-10 production in response to TNF-α.
Conclusion
The novel heterozygous mutation p.M37K in IκB-α impairs NF-κB activation causing autosomal dominant EDA-ID with an expanded clinical phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) is a rare syndrome characterized by partial or complete absence of exocrine sweat glands, sparse hair growth and hypo- or anodontia, conical teeth, associated with severe bacterial and viral infections and defective Immunoglobulin (Ig) production [1, 2]. EDA-ID is caused by mutations in genes encoding proteins involved in the activation of Nuclear Factor κB (NF-κB), a transcription factor pivotal for innate and adaptive immune responses, cell adhesion, cell growth, apoptosis, and ectodermal development [3–5]. The majority of patients with this disorder have the X-linked form (XL-EDA-ID) caused by hypomorphic hemizygous mutations in the gene encoding the Nuclear Factor κB Essential Modulator (NEMO; also known as IKBKG; OMIM: *300248), the regulatory subunit of the IκB kinase (IKK) complex, IKK-γ. In addition, few patients with autosomal dominant inheritance (AD-EDA-ID) caused by hypermorphic mutations in the gene encoding NF-κB inhibitor IκB-α (NFΚBIA; also known as IΚBΑ; OMIM *164008) have been described [6–10]. Whereas anhidrotic ectodermal dysplasia, defective Ig production, and severe bacterial infections are associated with both forms of EDA-ID, patients with AD-EDA-ID additionally show marked lymphocytosis and significant, albeit variable, T cell immunodeficiency [11].
The NF-κB transcription factor, composed of members of the Rel family proteins (including p50,and p65 [RelA]), is ubiquitously expressed and binds DNA as a homo- or heterodimer after cell activation by a variety of stimuli including pro-inflammatory cytokines, Interleukin (IL)-1, tumor necrosis factor (TNF)-α, bacterial lipopolysaccharide (LPS), viral proteins or stress. In resting cells, the NF-κB dimers are present in the cytoplasm, retained there by association with inhibitory κB proteins (IκB) such as IκB-α, IκB-ß and IκB-ε. Upon stimulation IκB-α is phosphorylated on specific serine residues S32 and S36 by the IKK complex consisting of the main proteins IKK-α, IKK-β and IKK-γ (NEMO), which triggers IκB-α polyubiquitination, and subsequent degradation by the 26S proteasome. This unmasks the nuclear localization signals on the NF-κB subunits allowing the dimeric transcription factor to translocate to the nucleus where it regulates expression of target genes [12–15].
To date, only six patients from five unrelated families have been described with IKBA mutations. Three of these patients show heterozygous missense mutations affecting the critical phosphoserine residue at position 32 (p.S32I) [6, 8] and three show nonsense mutations causing premature termination codons at positions 9 (p.Q9X), 11 (p.W11X), and 14 (p.E14X) [7, 9, 10]. In the latter two mutations, translation of the protein reinitiates at the next ATG (Methionine) which is in-frame at position 37. This generates a truncated IκB-α protein lacking the first N-terminal 36 amino acids containing both phosphoserine residues (S32 and S36). In the p.Q9X mutation, translation of the protein reinitiates at two ATG codons (M13 and M37) resulting in N terminus–truncated IκB-α proteins, wherein one (IκB-α ∆1–36) lacks both serine phosphorylation sites. The four unique mutations reported to date result in a protein that is resistant to degradation and lead to impaired NF-κB activation [6, 7, 9, 10].
We report here a child with AD-EDA-ID phenotype who, in addition, developed hypothyroidism, hypopituitarism, recurrent candidiasis, and was found to have low IL-17 T cell counts. While NEMO was found to be wild type, we identified a novel missense mutation in IκB-α (p.M37K) and were able to demonstrate that this heterozygous amino acid substitution interferes with the degradation of IκB-α resulting in a gain-of-function IκB-α protein impairing NF-κB activation.
Patient, Material and Methods
Case Report
The male patient was born to non-consanguineous healthy Caucasian parents after an uncomplicated pregnancy. At 6 months of age, he started to suffer from recurrent bacterial respiratory infections (mostly Haemophilus influenzae), chronic mucocutaneous candidiasis (CMC), chronic diarrhea, and failure to thrive. By 2 years of age, he developed signs of EDA including hypodontia and conical teeth, sparse hair, dry and rough skin with papulous exanthema (Fig. 1).

Clinical features of the patient with AD-EDA-ID. a dry and rough skin with papulous exanthema, b sparse hair, c conical teeth
Routine laboratory evaluation showed persistent elevated white blood cell counts (WBC) with relative and absolute lymphocytosis. At the age of 7 months, serum IgM reached 600 mg/dl (normal range 40–180 mg/dl), whereas IgG (<60 mg/dl; normal range 331–1200 mg/dl) and IgA (5.1 mg/dl; normal range 21–188 mg/dl) were found considerably below normal range. CD40 ligand expression on activated CD4+ T cells was normal. While fully immunized, no specific IgG-antibodies were detectable against diphtheria, tetanus, H. influenzae type b and Streptococcus pneumoniae. At 2.5 years of age, he was started on subcutaneous Ig (SCIG) therapy, cotrimoxazole as Pneumocystis jiroveci prophylaxis, and fluconazole for CMC with excellent results. However, at 4 years of age, he developed H. influenzae pneumonia and his condition deteriorated. Basal plasma cortisol levels were undetectable (<0.1 μg/dl), FT4 was decreased (0.7 ng/dl (normal 0,8–1,8 ng/dl)) and TSH moderately elevated (17 μU/ml (normal 0,85–6,0 μU/ml); those values had been within normal range in routine samples obtained earlier (13.2 μg/dl, 1.07 ng/ml and 2.1 μU/ml, respectively). Ultrasound diagnostic studies are compatible with autoimmune thyroiditis (Electronic supplementary material, Figure A1) although anti-thyroid antibodies could not be detected. Subsequent endocrinologic tests revealed hypothalamic hypopituitarism (growth hormone stimulation test with arginine was abnormally low, growth hormone releasing hormone (GHRH) test was normal with a pronounced growth hormone increase after GHRH application). Magnetic resonance imaging of the brain showed an undersized pituitary gland compatible with hypopituitarism (Electronic supplementary material, Figure B1 and C1). Anti-hypothalamic antibodies were not detected in the patient’s serum by immunohistochemistry (method described in Electronic supplementary material) [16]. L-thyroxin and hydrocortisone substitution therapy was started.
Because of increasing frequency of severe infections despite SCIG therapy and antibiotic prophylaxis, hematopoietic stem cell transplantation (HSCT) for the correction of the immunodeficiency was considered (Dupuis-Girod, Cancrini et al. 2006). At 4 9/12 years of age he received unmanipulated bone marrow from an HLA-matched (10/10) unrelated donor after reduced toxicity conditioning with treosulfan (42 g/m2), fludarabine (150 mg/m2) and alemtuzumab (0.9 mg/kg). Graft versus host disease (GVHD) prophylaxis comprised cyclosporine A and mycophenolate mofetil, which were discontinued on days +140 and +28, respectively. The patient developed acute GVHD Grade II (skin and liver) on day +30 followed by chronic GVHD Grade I (quiescent onset) starting on day 175 affecting the liver which was successfully treated with methylprednisolone for 30 days. Chimerism analysis of peripheral blood demonstrated 100 % donor cells until day +100 with a slow decrease to a stable level of 80–90 % donor cells at 15 months post transplantation. More than 1 year after HSCT, the patient was doing well and remained without severe infections. He continued to have chronic diarrhea (improved compared to before HSCT), candidiasis of the oral cavity requiring fluconazole treatment which remained until about 12 months post HSCT and minimal eczema requiring no treatment. His immunologic reconstitution 12 months after HSCT was slightly delayed with normalization of T-, B-, and NK- cell counts (except for low CD8+ cells), normalization of IL-17 T cell counts and mitogen and antigen stimulation in vitro, but with sustained low memory B cells (Table 1). Therefore, the patient continued to receive SCIG and antibiotic prophylaxis. Without remarkable pathologic findings during regular post-transplant follow-up and without central line placement or immunosuppressive treatment for more than 12 months, he unexpectedly died from overwhelming sepsis and purulent pericarditis due to Enterococcus faecium and Acinetobacter lwoffii 18 months post HSCT.
Immunophenotyping and Lymphocyte Proliferation
The study was approved by the local ethical review board. Written informed consent for blood and skin biopsy samples, and molecular genetic testing was obtained according to the requirements of the review board. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood using Ficoll-Paque PLUS (Bioscience AB, Uppsala, Sweden) and phenotyped by multicolor flow cytometry with the following antibodies (BD Biosciences, San Jose, California, USA): anti-CD19, anti-CD10, anti-CD24, anti-CD27, anti-CD38, anti-IgM, anti-IgD, anti-CD4, anti-CD8, anti-CCR7, anti-CD62L, anti-CD27, anti-CD45RA, anti-CD45RO, anti-CD25, anti-CD127, and anti-FOXP3 on a FACSCalibur flow cytometer (BD Biosciences) and analyzed with CellQuestPro software (BD Biosciences). IL-17 producing T cells were identified by means of intracellular staining of CD4+ T cells for the production of IL-17, as previously described [17]. Lymphocyte proliferation assays were performed according to a standard protocol [18]. Before harvesting, cells were pulsed with [3H]thymidine for 18 h and incorporation of [3H]thymidine was evaluated by liquid scintillation counting.
Cytokine Analysis in Response to NF-κB Activators
Cytokine production was evaluated in whole blood as previously described [19]. Cells were stimulated with IL-1ß (20 ng/ml; R&D Systems Europe, Lille Cedex, France), TNF-α (20 ng/ml; R&D Systems Europe), LPS from Salmonella Minnesota (TLR4 ligand 1 ng/ml; Sigma-Aldrich Chimie SARL, Lyon, France), heat killed Staphylococcus aureus Cowan I (SAC) (5 × 106 particles/ml; Invivogen, San Diego, California, USA), Pam2CSK4 (TLR2/6 ligand, 10 μg/ml; Invivogen), and PMA/Ionomycin (10−7 M and 10−5 M, respectively; Sigma-Aldrich) for 48 h. Supernatants were collected and the secretion of IL-10 and IL-6 were measured using specific ELISA (Sanquin, Mast Diagnostic, United Kingdom) kits according to the manufacturer’s instructions.
Genetic Analysis
The genes IKBKG (OMIM: *300248) and IKBA (OMIM: *164008) were amplified from genomic DNA (gDNA) and cDNA by polymerase chain reaction (PCR) as previously described [20] using specific oligonucleotide primers (specific conditions and primer sequences available upon request). Briefly, gDNA was prepared from whole blood by using the QIAampDNABlood Mini Kit (QIAGEN, Valencia, California, USA) and cDNA was synthesized from total mRNA extracted from 5 × 106 fresh PBMCs with Trizol (Invitrogen, Carlsbad, California, USA) with the Omniscript RT Kit (QIAGEN). The amplified gene fragments were sequenced with the ABI Big Dye Terminator mix (Applied Biosystems, Foster City, California, USA) and analyzed with a 3730xl DNA Analyzer (Applied Biosystems). The observed sequences were compared with those from GenBank. To predict the consequence of the novel mutation p.M37K in the IκB-α protein we used the bioinformatic methods MutationTaster [21], Polyphen-2 and SIFT to evaluate disease-causing potential of sequence alterations by using physical and comparative considerations.
Cell Culture
HeLa cells were cultured in Dulbecco’s-modified Eagle’s medium (DMEM, Invitrogen Live Technologies, Grand Island, New York, USA) supplemented with 10 % heat inactivated fetal bovine serum (FBS, Media Tech, Manassas, Virginia, USA). A total of 2.5 × 106 cells/well were maintained in 6-well plates in a final volume of 2 ml DMEM/10 % FBS and incubated at 37 °C for 24 h before transfection. Human fibroblasts were cultivated in DMEM plus 15 % FBS until 80 % confluence; following trypsinization approximately 2 × 105 cells/well were transferred to 6-well plates, maintained in a total volume of 2 ml medium and incubated at 37 °C for 48 h before stimulation. Prior to the treatment medium was changed to DMEM containing 15 % human serum.
Expression Plasmids
The wild type (WT) human IKBA cDNA was purchased from OriGene (SC320207, nm020529; OriGene Technologies, Rockville, Maryland, USA) and the mutants S36A-IκB-α (a mutation never observed in a patient but demonstrated from in vitro experiments to have the same effect on IκB-α activity as the S32I mutations reported in AD-EDA-ID patients) and M37K-IκB-α were created using the Stratagene QuickChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, California, USA) according to the protocol recommendations. The IKBA cDNA constructs were amplified by PCR and appropriate primers with recombination sites providing an efficient substrate for Gateway Cloning Vectors (Invitrogen Life Technologies). Gateway Cloning was performed as previously described with slight modifications [22] using a pDONR221 donor vector and a pcDNA3.1/nV5-DEST vector (Invitrogen Life Technologies) containing a N-terminal V5 epitope tag to create an expression vector encoding for the wild type or the two mutant IKBA alleles.
Transfections
The (V5-tagged) cDNA expression vectors described above or an empty control vector (pcDNA3.1-nV5) were either transfected alone or together with a NF-κB luciferase reporter vector and a Renilla reporter vector into HeLa cells. Before transfection the culture medium was changed to DMEM only. A total of 1 μg (if not otherwise indicated) of plasmid DNA were mixed with Fugene HD (Roche Indianapolis, Indiana) in a 3:1 ratio (3 μl Fugene: 1 μg DNA) in a total volume of 100 μl DMEM. After incubation for 20 min at room temperature the transfection mix was added drop wise to confluent cells grown in 6-well plates. For IκB-α degradation assays, the cells were incubated for 24 h before stimulation and for luciferase assays, protein expression was allowed for 16 h prior to stimulation. Before stimulation the culture medium was changed to fresh DMEM only.
IκB-α Degradation Through TNF- and Toll-Like- Receptor Signaling in Patient and Control Fibroblasts
To determine the effect of the mutation on TLR signaling, IκB-α degradation was tested in patient and control fibroblasts by flow cytometry. Briefly, fibroblasts were stimulated with TNF-α (20 ng/ml), LPS (1 μg/ml, TLR4 agonist) and Pam3CSK4 (1 μg/ml, TLR1/2 agonist) for 30 min at 37 °C or left untreated. The cells were fixed and permeabilized (Cytofix/Perm Buffer II, BD Biosciences) and stained with PE labeled anti-IκB-α antibody (Mouse IgG1) (BD Biosciences). After 30 min of incubation in the dark, the cells were resuspended in filter-sterilized staining buffer (DPBS + 2 % HI-FBS + 0.09 % sodium acid) and analyzed by flow cytometry on a LSR II flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (TreeStar Inc., Ashland, Oregon, USA)
Whole Cell Extracts and Western Blot Analysis of Transfected HeLa Cells
For IκB-α degradation assays, transfected HeLa cells were stimulated with TNF-α (20 ng/ml) at indicated time points. After washing the adherent cells twice with ice cold PBS, cells were collected by scraping and centrifuged at 2500 rpm for 5 min at 4 °C to pellet cells. Whole cell extracts were prepared using M-PER extraction buffer (Pierce, Rockford, Illinois) with Protease Inhibitors (1:100) (Pierce) and Halt Protease Inhibitor Cocktail (1:100) (Pierce). After incubation for 10 min at 4 °C under gentle rotation, extracts were cleared by centrifugation at high speed for 15 min at 4 °C. Protein concentration in supernatants was determined by Bradford assay. 10 μg of proteins were separated by standard SDS-PAGE and transferred to western blot membranes. Blots were stained with murine Anti-V5-horseradish peroxidase (HRP)-conjugated monoclonal antibody (mAb) (Invitrogen Life Technologies) at a dilution of 1:5000 to detect the presence of V5-tagged IκB-α protein. As a control for equal protein loading, blots were stained with a murine anti α-Tubulin mAb (Sigma, Saint Louis, Missouri) and secondary HRP-conjugated polyclonal anti mouse IgG antibody (Biosource-Invitrogen, Carlsbad, California).
Electrophoretic Mobility Shift Assay (EMSA) Using Transfected HeLa Cells
Nuclear extracts of HeLa cells expressing exogenous M37K-, S36A- or WT-IκB-α-, or an empty control vector (pcDNA3.1-nV5) were evaluated for the presence of p50/p65 dimers after stimulation with TNF-α (20 ng/ml) by electrophoretic mobility shift assay (EMSA) using an NF-κB-specific labeled oligonucleotide binding-site probe derived from the c-Fos promoter [23]. Briefly, 24 h following transfection, the cells were stimulated with TNF-α for 30 min and nuclear extracts were prepared as previously described [24]. 5 μl of nuclear extract were mixed with 1 μl poly dI/dC (1 mg/ml) and incubated with 2 μl of a 32P labeled double stranded oligonucleotide probe containing a NF-κB binding site [25] and 12 μl sterile water for 25 min at room temperature. After adding 2 μl of EMSA dye, samples were run on a 30 % acrylamide gel in 0.5 × Tris base, boric acid and EDTA (TBE) buffer.
Luciferase Assays
HeLa cells were cotransfected with 0.5 μg of the firefly luciferase expression vector containing a NF-κB responsive element (pNF-κB(1); Panomics, Fremont, California, USA), 0.5 μg of the pRL-TK renilla luciferase control vector (Promega, Madison, Wisconsin, USA), and 0,1 μg of the indicated IκB-α expression vectors or empty control vector. 16 h after transfection cells were stimulated with TNF-α (10 ng/ml) for 4 h. Luciferase assays were performed by using the Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer’s instructions. Luminescence was determined in duplicate using a VICTOR3™ V Multilabel Counter 1420 luminometer (PerkinElmer, San Jode, California) with autoinjectors. Data were analyzed for statistical significance using the unpaired t-test and GraphPad PRISM 4.03 software (GraphPad Software, San Diego, California, USA). A P value of <.05 was considered significant.
Results
Cellular Phenotype Before HSCT
The lymphocyte subpopulations consistently displayed high CD4+ and low CD8+ T cell counts leading to a high CD4/CD8 ratio (range 5–7), and normal proportions of B cells (Table I). Naive (CD45RA) T lymphocyte counts were high, while those of memory (CD45RO) T lymphocytes were low resulting in a markedly decreased ratio of CD45RO/CD45RA T cells. IL-17 producing T cells and γ/δ T cells were reduced, and CD4+CD25+Foxp3+ T regulatory cells (Tregs) in the lower normal range. B-cell subpopulations revealed reduced total CD19+CD27+ memory cells and absent CD19+CD27+IgM−IgD− cells (switched memory B cells). Proliferation in response to anti-CD3 (OKT3) and Staphylococcus aureus Cowan I (SAC) was low, whereas proliferation in response to mitogens (phytohemagglutinin, pokeweed mitogen and Concanavalin A) and antigens (including a mix of diphtheria/tetanus/Streptolysin O/mumps antigens) was normal (Table I). We then tested the IL-6 and IL-10 production by the patient’s whole blood cells in response to PMA/Ionomycin, TLR ligands, IL-1ß, TNF-α, and SAC. The IL-6 production was reduced in response to IL-1β, Pam2CSK4 (TLR2/6 agonist), LPS (TLR4 agonist) and PMA/Ionomycin, while it was normal in response to SAC (Fig. 2a). IL-10 production in response to TNF-α was absent (Fig. 2b). These results suggested a genetic defect in the NF-κB signalling pathway.

Impaired secretion of IL-6 and IL-10 upon NF-κB dependent stimuli. Whole blood cells from the patient and a healthy control were stimulated with (a) medium only, IL-1β, SAC, LPS, PAM2CSK4 (Pam2) or PMA/Ionomycin, showing reduced IL-6 concentration in patient supernatant after 48 h co-culture with LPS and PMA/Ionomycin. b Patient’s IL-10 concentration in the supernatant was reduced after TNF-α stimulation for 48 h compared to a normal control. Results are provided as means ± SEM of two independent experiments. PMA, Phorbol 12-myristate 13-acetate
Novel Mutation p.M37K in IκB-α
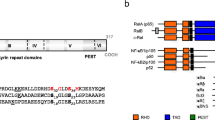
Based on clinical and immunologic data compatible with a genetic defect in the NF-κB signalling pathway, we sequenced the IKBKG gene without identifying a mutation (data not shown). We then considered IKBA sequencing and found a novel heterozygous c.110 T > A (according to NCBI Reference Sequence: NM_020529.2) point mutation resulting in an amino acid substitution from methionine to lysin at position 37 of the IκB-α protein (Fig. 3a). This mutation is unique and does not affect one of the serine residues at position 32 and 36 critical for phosphorylation and degradation of IκB-α and has not, to our knowledge, been previously described (Fig. 3b). Using the UCSC Genome Bioinformatics database (http://genome.ucsc.edu), we found that the methionine at position 37 of IκB-α is localized in a core of amino acids highly conserved among different species (Fig. 3c). Furthermore, MutationTaster analysis predicts the p.M37K variant to be a pathogenic mutation and no single nucleotide polymorphism was found in the altered region using the MutationTaster and Ensembl database (http://www.ensembl.org). Using Polyphen-2 and SIFT bioinformatic analysis, the mutation is predicted to be probably damaging with a score of 0.971 (sensitivity: 0.77; specificity: 0.96) and predicted to affect protein function with a score of 0.00 (intolerant), respectively. The mother of the patient carried the wild type IKBA on both alleles; DNA from the healthy father was not available.

NFKBIA/IKBA mutation analysis. a Chromatogram showing a normal control sequence and the heterozygous point mutation (c.110 T > A; p.M37K) identified in the patient. b Protein structure of IκB-α indicating the two phosphoserine sites (S32 and S36), the six ankyrin repeat domains, the PREST domain, the previously reported mutations (black), and the novel M37K mutation identified in our patient (red). The * indicates the number of patients with these known mutations identified to-date. c IKBA sequence alignment demonstrating a conserved methionine at position 37 (red box) among different species
p.M37K Prevents IκB-α Degradation
The identification of a novel, potentially disease-causing mutation in the patient led us to investigate the functional consequences of this mutation. We first analyzed IκB-α degradation in primary fibroblasts of the patient and a normal control after stimulation with TLR1/2 (PAM3CSK4) and TLR4 (LPS) agonists and TNF-α. The patient’s fibroblasts failed to degrade IκB-α in response to all three stimuli while the healthy control fibroblasts showed normal IκB-α degradation (Fig. 4a). We then confirmed defective degradation of IκB-α in transfection experiments. HeLa cells carrying the IκB-α-WT protein showed degradation after 20 min of stimulation, whereas almost no decrease in the level of IκB-α was seen in the cells expressing the IκB-α-M37K mutant, similar to the IκB-α-S36A mutant (Fig. 4b).

Defective IκB-α degradation. a Primary fibroblasts derived from the patient and a healthy control were analyzed for IκB-α degradation by flow cytometry after stimulation with TNF-α, LPS (TLR4 agonist) and PAM3CSK4 (TLR1/2 agonist). While all three stimuli induce IκB-α degradation in control fibroblasts, they fail to induce degradation in the patient’s fibroblasts, indicating defective TLR signaling. b IκB-α degradation in transfected HeLa cells after stimulation with TNF-α for indicated time points; while degradation of wild type IκB-α at 20 min is almost complete, no degradation was observed with both mutant constructs (IκB-α-S36A and IκB-α-M37K). The IκB-α protein generated by the transfectant was detected using a specific antibody against the V5 epitope. One of three independent experiments is shown
p.M37K Leads to Reduced Nuclear Translocation of NF-κB
Since the p.M37K mutant prevents IκB-α degradation keeping the nuclear localization signals on the NF-κB subunits masked, we hypothesized that nuclear translocation of NF-κB is reduced. The levels of NF-κB p50/p65 dimers binding to DNA after stimulation with TNF-α were similar in non transfected HeLa cells, HeLa cells transfected with empty vector, and the IκB-α-WT. In contrast, HeLa cells transfected with IκB-α-S36A and IκB-α-M37K showed decreased amounts of NF-κB p50/p65 dimer binding to the nuclear DNA (Fig. 5a).

Reduced NF-κB translocation to the nucleus and reduced NF-κB dependent gene transcription. a EMSA assay to evaluate defective NF-κB activation was performed. Reduced amounts of nuclear DNA-binding NF-κB dimers (p50/p65) in both IκB-α-S36A and IκB-α-M37K mutant HeLa cell constructs after stimulation with TNF-α was detected. One of two independent experiments is shown. b Luciferace assays to evaluate effect of IκB-α mutation on NF-κB activation. Fold induction obtained from Luciferase activity was calculated from stimulated reporter activity (firefly/renilla ratio) divided by the reporter activity of unstimulated cells. Mean values of three independent experiments are shown. Luciferase activity was significantly reduced (p < 0.05, unpaired t-test) in HeLa cells transfected with the IκB-α-S36A and IκB-α-M37K mutant compared to cells transfected with IκB-α-wild type or an empty expression vector (control)
To prove that the mutated IκB-α protein is able to block gene transcription, we used a NF-κB luciferase reporter gene assay. HeLa cells expressing exogenous IκB-α wild type achieved a 28 fold up-regulation of luciferase activity in response to stimulation with TNF-α while the up-regulation of luciferase activity was significantly impaired (64 % decrease compared to the WT) in both IκB-α-mutated constructs showing only a 10 fold increase (Fig. 5b). This indicates that the mutated protein IκB-α-M37K, similar to the IκB-α-S36A impairs gene expression controlled by NF-κB activation and has a dominant negative effect.
Discussion
We report a young boy with EDA-ID, who presented with presumed autoimmune thyroiditis and hypopituitarism, recurrent candidiasis, hyper IgM phenotype, low normal Treg and low IL-17 T cell counts. We identified a novel heterozygous IKBA missense mutation (p.M37K) that leads to defective IκB-α degradation impairing NF-κB activation as demonstrated by reduced NF-κB nuclear translocation and NF-κB dependent gene transcription. These findings are consistent with those previously described for two different IKBA mutations p.S32I and p.E14X in AD-EDA-ID patients leading to proteins resistant to ligand-induced degradation and as a consequence, inhibition of NF-κB activation. [6–8]. In addition, the recently described mutation p.Q9X results in a N terminal-truncated IκB-α protein showing a significant dose-dependent inhibitory effect on NF-kB reporter gene activity in vitro [10]. Moreover, previously published results in which IκB-α cell constructs bearing mutations of serine 36 (S36A) or preceding amino acids (D31A/D35A) result in degradation resistant IκB-α proteins and reduced NF-κB activation [12, 13, 26, 27] are in agreement with our findings. Impaired NF-κB function was confirmed in the patient reported here by demonstrating reduced IL-6 and IL-10 production by whole blood leukocytes in response to NF-κB dependent stimulation. The discrepant result of normal IL-6 production by SAC stimulation is in accordance with the observation that antigen presenting cells, which are the main producers of IL-6, from XL-EDA-ID patients and presumably AD-EDA-ID patients, retain the capacity to respond to SAC stimulation [28]. This can be explained by the fact that the IL-6 promoter includes a number of control regions which can be synergistically transactivated by different transcription factors in addition to NF-κB, including C/EBP, c-Jun and AP-1 [29].
Our patient presented with typical clinical findings of AD-EDA-ID including conical teeth, sparse hair, dry skin, recurrent bacterial infections, CMC and chronic diarrhea. However, to our knowledge, this is the first patient with IKBA mutation with hypothyroidism and hypothalamic hypopituitarism. Since hypopituitarism is a common feature of many chronic disorders and most often does not reflect an autoimmune etiology [30–33], we can only speculate that a cell mediated autoimmune mechanism is the basis of the polyendocrinopathy in our patient, especially since anti-thyroid or anti-rodent hypothalamus autoantibodies could not be identified. A developmental defect as cause of the hypothalamic hypopituitarism is less likely, since the thyroid hormone and cortisol levels had been normal at a younger age. Autoimmune manifestations such as enterocolitis are frequently reported in primary immunodeficiencies (PID) including XL-EDA-ID and AD-EDA-ID [34–36] and the underlying mechanisms of the homeostasis between activation and suppression of immune responses in PID have been recognized. NF-κB is required for the development of both anti-inflammatory and pro-inflammatory cytokines [37] which corroborates with our findings of simultaneously defective IL-10 and IL-6 production by patient’s leukocytes. Moreover, NF-κB is required for the development of regulatory T cells (Tregs) in mice [37] and defective development of Tregs is the direct cause of autoimmune manifestations in patients with IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome [38]. While Tregs have not yet been systematically examined in EDA-ID patients, mice with T cells lacking NEMO exhibit impaired formation of Treg cells [39, 40]. While Treg cell numbers were in the lower normal ranges, we did not measure Treg function in our patient.
Because the NF-κB transcription factor family plays an important role in B- and T-cell proliferation, activation, cytokine secretion and class switch recombination during B cell maturation [41, 42], it is not surprising that defective NF-κB signalling in EDA-ID leads to significant abnormalities in B and T cell development [11]. The patient’s abnormal immunologic parameters, including high CD4+ and low CD8+ T cell subsets, decreased CD45RO memory T cells and a hyper-IgM B cell phenotype with lack of switched memory B cells and defective antibody response, were similar to those previously reported in patients with IKBA mutation [6, 8, 9, 36].
Decreased T cell proliferation in response to anti-CD3 monoclonal antibody was previously observed in patients with IKBA mutations [6]. NF-κB has been described as an important transcription factor in pro-inflammatory immune responses including the development of IL-17 T cells [37]. Low IL-17 T cell counts, despite high CD4+ T cell numbers observed in our patient have not been previously reported in AD-EDA-ID, broadening the degree of defective T cell development in patients with IKBA mutation [6–9]. IL-17 T cells play a prominent role in controlling infections against extracellular bacteria and fungi in mice [43] and human CMC is associated with reduced numbers of IL-17 T cells in a number of PIDs including the AD-hyper-IgE syndrome due to STAT3 mutations and CMC due to STAT1 gain-of-function mutations [17, 20, 44–47], or mutations in IL-17 or its receptor [44]. It is reasonable to assume that the low IL-17 T cell count in our patient is caused by impaired NF-κB activity due to the M37K mutation in IKBA, leading in turn to susceptibility to CMC. It is, however, possible that the susceptibility to CMC could have been caused by abnormalities in other defense systems dependent of the NF-κB pathway, such as C-type lectin pattern recognition receptors including dectin-1 and dectin-2 [48]. In accordance, defects in dectin-1 signaling have been associated with CMC [49, 50].
Hematopoietic stem cell transplantation for patients with a defect in the NF-κB pathway has been carried out with conflicting results [7, 51, 52]. Based on the success of HSCT in at least a few patients with IKBKG or IKBA mutation and the severe clinical phenotype of our patient, we performed allogeneic HSCT from a matched unrelated donor following reduced intensity conditioning. Although he developed mild acute and chronic GVHD, he remained well without severe infections for one and a half years after transplantation. His immune reconstitution was slightly delayed and B cell maturation incomplete, possibly due to mild chronic GVHD. He unexpectedly died from overwhelming sepsis and bacterial pericarditis 18 months after transplantation caused by gastrointestinal and commensal bacteria, which might be related to dysfunctional mucosal barrier. Disruption of the epithelial barrier causing severe infection with enteral bacteria after successful immune reconstitution post transplantation has been observed in patients with IKBKG mutations suggesting a critical role for NF-κB signaling in intestinal epithelial barrier function [52, 53]. While this highlights the possibility that post-transplant complications may impact the outcome of HSCT in this syndrome, it is important to point out that the patient significantly improved clinically and immunologically after transplantation and that HSCT may cure the cellular immunodeficiency consistently present in patients with AD-EDA-ID phenotype.
In summary, our immunologic, molecular and functional data demonstrate an essential role for amino acid M37 in IκB-α degradation and that the heterozygous p.M37K mutation is capable of blocking NF-κB activation due to gain-of-function of the IκB-α protein.
Abbreviations
- AD-EDA-ID:
-
Autosomal dominant ectodermal dysplasia with immunodeficiency
- CMC:
-
Chronic mucocutaneous candidiasis
- EDA-ID:
-
Ectodermal dysplasia with immunodeficiency
- GHRH:
-
Growth hormone releasing hormone
- GVHD:
-
Graft versus host disease
- HSCT:
-
Hematopoietic stem cell transplantation
- IKBA:
-
NF-κB inhibitor IκB-α
- IPEX:
-
Immunodysregulation, polyendocrinopathy, enteropathy, X-linked
- NEMO:
-
Nuclear Factor κB Essential Modulator
- NF-κB:
-
Nuclear Factor κB
- PID:
-
Primary Immunodeficiency
- TLR:
-
Toll like receptor
- Tregs:
-
T regulatory cells
- SCIG:
-
Subcutaneous immunoglobulin
- XL-EDA-ID:
-
X linked form of ectodermal dysplasia with immunodeficiency
References
Abinun M. Ectodermal dysplasia and immunodeficiency. Arch Dis Child. 1995;73:185.
Carrol ED, Gennery AR, Flood TJ, Spickett GP, Abinun M. Anhidrotic ectodermal dysplasia and immunodeficiency: the role of NEMO. Arch Dis Child. 2003;88:340–1.
Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41.
Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85.
Courtois G. The NF-kappaB signaling pathway in human genetic diseases. Cell Mol Life Sci. 2005;62:1682–91.
Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, Dupuis-Girod S, Bodemer C, Livadiotti S, Novelli F, Rossi P, Fischer A, Israel A, Munnich A, Le Deist F, Casanova JL. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Investig. 2003;112:1108–15.
Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, Quinones R, Gelfand EW, Jain A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29:861–8.
Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, van de Vosse E, van Tol M, Bredius R, Ottenhoff TH, Weemaes C, van Dissel JT, Lankester A. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. The J Exp Med. 2004;200:559–68.
McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. The J Allergy Clin Immunol. 2007;120:900–7.
Ohnishi H, Miyata R, Suzuki T, Nose T, Kubota K, Kato Z, Kaneko H, Kondo N. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. The J Allergy Clin Immunol. 2012;129:578–80.
Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. 2011;24:490–7.
DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–304.
Sun S, Elwood J, Greene WC. Both amino- and carboxyl-terminal sequences within I kappa B alpha regulate its inducible degradation. Mol Cell Biol. 1996;16:1058–65.
Chen CL, Yull FE, Kerr LD. Differential serine phosphorylation regulates IkappaB-alpha inactivation. Biochem Biophys Res Commun. 1999;257:798–806.
Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-kB activity. Annu Rev Immunol. 2000;18:621–63.
Adams JC. Biotin amplification of biotin and horseradish peroxidase signals in histochemical stains. J Histochem Cytochem. 1992;40:1457–63.
Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. The J Allergy Clin Immunol. 2008;122:181–7.
Maluish A, Strong D. Lymphocyte proliferation. In: Rose N, Friedman H, Fahey J, editors. Manual of clinical laboratory immunology. Washington: American Society for Microbiology; 1986. p. 274–81.
Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, Doffinger R, Bonnet M, Filipe-Santos O, de Beaucoudrey L, Durandy A, Horneff G, Novelli F, Wahn V, Smahi A, Israel A, Niehues T, Casanova JL. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701.
Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, Rieber N, Cremer R, Maass E, Dopfer R, Reichenbach J, Wahn V, Hoenig M, Jansson AF, Roesen-Wolff A, Schaub B, Seger R, Hill HR, Ochs HD, Torgerson TR, Belohradsky BH, Renner ED. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. The J Allergy Clin Immunol. 2010;126:611–7.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, Ziegler SF. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol. 2006;177:3133–42.
Torgerson TR, Colosia AD, Donahue JP, Lin YZ, Hawiger J. Regulation of NF-kappa B, AP-1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-kappa B p50. J Immunol. 1998;161:6084–92.
Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419.
Cordle SR, Donald R, Read MA, Hawiger J. Lipopolysaccharide induces phosphorylation of MAD3 and activation of c-Rel and related NF-kappa B proteins in human monocytic THP-1 cells. J Biol Chem. 1993;268:11803–10.
Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15:2809–18.
Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–83.
Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–8.
Faggioli L, Costanzo C, Donadelli M, Palmieri M. Activation of the Interleukin-6 promoter by a dominant negative mutant of c-Jun. Biochim Biophys Acta. 2004;1692:17–24.
Hana V, Kosak M, Masopust V, Netuka D, Lacinova Z, Krsek M, Marek J, Pecen L. Hypothalamo-pituitary dysfunction in patients with chronic subdural hematoma. Physiol Res. 2012;61:161–7.
Yuasa M, Fujiwara S, Oh I, Yamaguchi T, Fukushima N, Morimoto A, Ozawa K. Rapidly progressing fatal adult multi-organ langerhans cell histiocytosis complicated with fatty liver disease. J Clin Exp Hematop. 2010;52:121–6.
McCaffery TD, Nasr K, Lawrence AM, Kirsner JB. Severe growth retardation in children with inflammatory bowel disease. Pediatrics. 1970;45:386–93.
Green JR, O’Donoghue DP, Edwards CR, Dawson AM. A case of apparent hypopituitarism complicating chronic inflammatory bowel disease in childhood and adolescence. Acta Paediatr Scand. 1977;66:643–7.
Bussone G, Mouthon L. Autoimmune manifestations in primary immune deficiencies. Autoimmun Rev. 2009;8:332–6.
Orange JS, Levy O, Geha RS. Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kappaB activation. Immunol Rev. 2005;203:21–37.
Kawai T, Nishikomori R, Heike T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergol Int. 2012;61:207–17.
Ruan Q, Chen YH. Nuclear factor-kappaB in immunity and inflammation: the Treg and Th17 connection. Adv Exp Med Biol. 2012;946:207–21.
Chang X, Zheng P, Liu Y. FoxP3: a genetic link between immunodeficiency and autoimmune diseases. Autoimmun Rev. 2006;5:399–402.
Cheng LE, Kanwar B, Tcheurekdjian H, Grenert JP, Muskat M, Heyman MB, McCune JM, Wara DW. Persistent systemic inflammation and atypical enterocolitis in patients with NEMO syndrome. Clin Immunol. 2009;132:124–31.
Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israel A, Rajewsky K, Pasparakis M. Mature T cells depend on signaling through the IKK complex. Immunity. 2003;19:377–89.
Jain A, Ma CA, Lopez-Granados E, Means G, Brady W, Orange JS, Liu S, Holland S, Derry JM. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Investig. 2004;114:1593–602.
Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34.
Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–71.
Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–8.
Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The J Exp Med. 2008;205:1551–7.
de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. The J Exp Med. 2008;205:1543–50.
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6.
van der Meer JW, van de Veerdonk FL, Joosten LA, Kullberg BJ, Netea MG. Severe Candida spp. infections: new insights into natural immunity. Int J Antimicrob Agents. 2010;36 Suppl 2:S58–62.
Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, Jamal S, Manguiat A, Rezaei N, Amirzargar AA, Plebani A, Hannesschlager N, Gross O, Ruland J, Grimbacher B. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–35.
Kingeter LM, Lin X. C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell Mol Immunol. 9: 105–12.
Dupuis-Girod S, Cancrini C, Le Deist F, Palma P, Bodemer C, Puel A, Livadiotti S, Picard C, Bossuyt X, Rossi P, Fischer A, Casanova J. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–11.
Fish JD, Duerst RE, Gelfand EW, Orange JS, Bunin N. Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant. 2009;43:217–21.
Pai SY, Levy O, Jabara HH, Glickman JN, Stoler-Barak L, Sachs J, Nurko S, Orange JS, Geha RS. Allogeneic transplantation successfully corrects immune defects, but not susceptibility to colitis, in a patient with nuclear factor-kappaB essential modulator deficiency. The J Allergy Clin Immunol. 2008;122:1113–8. e1.
Acknowledgment
We thank the family for participating. For technical assistance and performing flow cytometry we thank Irmgard Eckerlein, Ottilie Bieberle, and Mayumi Hoffmann. This work was supported by the ESID long-term fellowship 2010 (to LFS), the Jeffrey Modell Foundation (to HDO), DFG RE2799/3-1 and the Fritz-Thyssen research foundation grant (Az. 10.07.1.159) (to EDR).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Lena F. Schimke and Nikolaus Rieber contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 141 kb)
Rights and permissions
About this article
Cite this article
Schimke, L.F., Rieber, N., Rylaarsdam, S. et al. A Novel Gain-of-Function IKBA Mutation Underlies Ectodermal Dysplasia with Immunodeficiency and Polyendocrinopathy. J Clin Immunol 33, 1088–1099 (2013). https://doi.org/10.1007/s10875-013-9906-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-013-9906-1