
Abstract
Background
The idea that the innate and adaptive immune systems are not separate entities is no longer new. In fact, it is surprising that this paradigm was accepted without question for so long. Many innate cells express cell surface molecules and soluble mediators that are essential for the development and activation of T cells and B cells. Yet among the innate cell populations, mast cells may play the major role in regulating adaptive immune cell function.
Discussion
This role first came to light in studies of mast cells and their involvement in the autoimmune disease experimental allergic encephalomyelitis, the major rodent model of multiple sclerosis and has subsequently been verified in many in vitro and in vivo model systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
What Are Mast Cells?
Like most cells of the immune system, mast cells derive from a pluripotent precursor cell present in the bone marrow and fetal liver where they acquire their lineage-specific features under the influence of stem cell factor (SCF) and interleukin (IL)-3 (for review, see [1]). Mast cells then leave the bone marrow and migrate to a wide variety of tissue sites including the thymus, lymph nodes, spleen, skin and heart, and the mucosa of the genitourinary, respiratory, and gastrointestinal tracts where they complete their differentiation and acquire tissue-specific characteristics. Within specific tissues, they are most often found adjacent to blood vessels and peripheral nerves. This distribution, particularly at sites most vulnerable to entry of infectious organisms, correlates with the potential of mast cells to act as a first line of defense in fighting infection [2]. Mast cells are normally defined by the cell surface expression of FcɛRI+ (the high-affinity IgE receptor) and c-kit+ (the SCF receptor), as well as the presence of granules that stain with metachromatic dyes such as toluidine blue.
Although mast cells do not have antigen-specific receptors that are strictly analogous to those present on B and T cells, the expression of a high-affinity IgE receptor on the mast cell surface endows these cells with similar abilities to respond in an antigen-specific way. The FcɛRI receptor binds monomeric IgE produced after a Th2-dominated antigen-specific response (reviewed in [3]). The tight association of IgE with this receptor poises the cell for immediate antigen response upon subsequent exposure to that antigen. Many of the downstream signaling pathways that lead to activation-induced changes in gene expression are reminiscent of T-cell receptor (TCR)- and B-cell receptor (BCR)-driven activation pathways. However, FcɛRI cross-linking not only elicits new gene expression similar to TCR- and BCR-mediated signals, but it can also trigger degranulation, a process whereby vesicles containing a myriad of pre-formed, pro-inflammatory mediators are released from the cell. Well-known mast cell products such as histamine, serotonin, chymase, and tryptase reside in these granules and can initiate the immediate hypersensitivity responses associated with allergies. Newly synthesized mediators include a multitude of cytokines, chemokines, prostaglandins, and leukotrienes that amplify the immediate type hypersensitivity response and contribute to late-phase allergic responses.
Mast Cells Act Outside the Realm of Allergy
There is no doubt that mast cells are major contributors to allergic responses. However, the extreme and exclusive focus on these cells in this setting probably impeded progress in our understanding of mast cells as major players in other inflammatory settings and perhaps in anti-inflammatory scenarios as well. A major breakthrough came with the discovery that antigen-IgE cross-linking through the FcɛRI is only one of many ways a mast cell can be activated. Agonists as diverse as complement, hormones, neuropeptides, cytokines, and microbial products that act through specific receptors are probably major mast cell stimuli in non-allergic disease settings. That Ig-independent modes of activation exist demonstrates that mast cells are not only functional after an adaptive response has been initiated when antibody-producing B cells are activated but that they also can act at the earliest stages of immune insult. For example, the microbial product receptors that are widely expressed on mast cells include the cell surface toll-like receptors as well as the intracellular nucleotide oligomerization domain (NOD) receptors [4–6]. During infections, these receptors trigger mast cells to express products that recruit other innate cells such as neutrophils and activate dendritic cells (see below). The critical protective role of mast cells in bacterial infections has been shown in a number of studies using gut or lung pathogens [7, 8].
Many of mast cells’ protective effects in bacterial infections can be attributed to the direct actions of mast cells on the microbes themselves or on the activation and recruitment of other innate cells. Yet there is now much evidence that mast cells also act on dendritic cells (DCs) and T cells (and B cells) to modulate their response through the inducible expression of a wide range of co-stimulatory/co-inhibitory molecules and cytokines that act on DCs, T cells, and B cells (for review, see [9–11]). We still do not understand the full range of molecules that are inducibly expressed by mast cells upon activation with Ig-independent agonists. Of interest is the observation that there are strain-specific and agonist-specific patterns of expression suggesting that mast cell responses and their effects on the adaptive immune response can vary depending on the setting [12, 13].
Studying Mast Cells In Vivo: Use of c-kit Mutant Mice
Much of the evidence for T cell–mast cell interactions has been generated using in vitro cell culture systems, but support for mast cell effects on adaptive responses also come from studies in in vivo models. The most common models utilize one of two strains of mast cell-deficient mice [14]. WBB6c-kitW/Wv (W/Wv) mice have two distinct mutations at the white spotting locus (W), the region that encodes the c-kit receptor. These mutations in combination do not obliterate c-kit signaling but reduce it to about 10% of wild-type levels. Because mast cells are exquisitely dependent on SCF for their development and maturation, this defect results in a virtual loss of all mast cells.
Another mast cell-deficient strain, Wsh, is superior in some respects because these mice have normal fertility and are not anemic as are W/Wv mice. However, these mice exhibit a time-dependent loss of mast cells—a delay that may make the reliable assessment of mast cell-dependent events in young (less than 12 weeks old) mice impossible. This could be a disadvantage in studies of type I diabetes in mice for example. The associated waves of β-cell death during pancreatic development that are thought to trigger disease occur early in the life of the diabetes-prone NOD mouse strain (fetus to <3 to 4 weeks of age) [15]. Such events could not be discerned as mast cell-dependent in Wsh mice because mast cells are still present in these animals at the time of the earliest inflammatory signals.
Mast cells can be restored to most tissues of W/Wv or Wsh mice by transfer of “bone marrow mast cells” derived from wild-type bone marrow after culture for 4–6 weeks in SCF and IL-3 [16]. These in vitro-derived committed mast cell precursors populate most tissues where mast cells are normal residents. If mast cells restore the phenotype in question, the mast cell-dependence of that phenotype is then confirmed.
Experimental Allergic Encephalomyelitis: A Murine Model of Multiple Sclerosis
Our laboratory uses W/Wv mice to study the role of mast cells in myelin oligodendrocyte glycoprotein (MOG)-induced experimental allergic encephalomyelitis (EAE). While there are many differences in EAE and multiple sclerosis (MS), the EAE model has been useful in defining some key events in the disease process that have led to therapeutics for MS [17, 18]. Similar to the human disease, EAE is dependent on the development of an autoreactive CD4+ T-cell response in the periphery and the subsequent migration of these and other inflammatory cells into to the central nervous system (CNS) [19]. The breach of the normally impermeable blood–brain barrier (BBB) is an important checkpoint in disease. Indeed, normal healthy individuals can have myelin-specific autoreactive T cells that remain circulating in the periphery and fail to breach the BBB [20].
Mast Cell-Deficient Mice Have Normal T-Cell Compartments
Because c-kit signaling may impact early T-cell development, it was necessary to determine whether the T-cell compartment in W/Wv mice is intact prior to studying T-cell-mediated disease in W/Wv mice. Thymic T-cell profiles (CD4, CD8 DN, DP, SP) are normal in W/Wv mice as are numbers of peripheral CD4+ and CD8+ T cells in the peripheral lymphoid organs [21]. Most significantly, the transfer of naïve wild type or W/Wv splenic T cells into a TCR-deficient mouse (with a normal mast cell compartment) followed by EAE induction results in equivalent disease irrespective of the source of T cells, confirming that there is no inherent defect in W/Wv T cells. That is, if primed in a mast cell-sufficient environment, W/Wv T cells can function normally.
Mast Cells Influence EAE Disease Severity
A comparison of EAE clinical scores revealed that W/Wv mice develop a significantly delayed disease course and with less clinical severity than their wild-type littermates [22]. Reconstitution of mast cells restores wild-type disease. Because mast cell-deficient mice can develop EAE disease symptoms under certain disease induction conditions, these cells are not required for disease. Rather, it is likely that mast cells’ major role is to amplify the inflammatory response. This role for mast cells has been verified in human disease as well. Gene expression profiles reveal histamine 1 receptor, tryptase, and FcɛRI transcripts are expressed in CNS plaques of MS patients [23].
Our original hypothesis proposed that mast cells act primarily in the CNS. This idea was prompted by reports that mast cells are present in the CNS, that mast cells can be directly activated by myelin, and that they express proteases that could contribute to damage of the myelin sheath [24, 25]. Surprisingly, although reconstitution with bone marrow-derived mast cells restores disease severity, this occurs in the absence of CNS re-population [26]. This observation does not eliminate the possibility that CNS mast cells have an important role in the effector phase of the disease. Indeed, the adoptive transfer of wild-type encephalitogenic T cells to wild-type and W/Wv recipients results in less severe disease in mast cell-deficient animals [21]. Thus, under conditions of identical peripheral T-cell priming, W/Wv mice are still resistant to severe disease. However, it does imply that mast cells are acting elsewhere. In view of the potential for mast cell–T cell crosstalk, we first chose to examine peripheral T-cell responses.
Mast Cells Act at Multiple Checkpoints Points in the Progression of EAE
In EAE, it is known that dendritic cells can internalize antigen at peripheral sites, process this antigen, and present it in the form of peptide fragments in association with major histocompatibility complex (MHC) molecules. The source of autoantigen in the periphery in MS is unknown but may be a microbial product that elicits a cross-reactive T- and B-cell response [27]. Antigen processing is accompanied by a series of events collectively termed “maturation” in which DCs acquire co-stimulatory molecules and migrate to draining lymph nodes. Initial T-cell priming occurs in the lymph nodes, and peak responses are observed several days following antigen exposure. Activated T cells migrate to the target tissue site (in the case of EAE/MS, the CNS) where they release cytokines/chemokines or carry out their cytolytic function. Other inflammatory cells are recruited, and the inflammatory response is amplified.
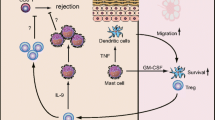
Mast Cell–Dendritic Cell Interactions
How might mast cells be activated in this setting, and how do these cells provide the optimal environment for T-cell responses? In the periphery, there are at least two possible stimulators of mast cells during disease induction, and both are related to the original immunogen. Myelin peptides are delivered subcutaneously to mice in complete Freund’s adjuvant. Both MOG35-55 and myelin basic protein peptides are known to cause degranulation of mast cells [28]. Complete Freund’s adjuvant contains Mycobacterium that can activate mast cells through toll-like receptors. Mast cells respond by releasing histamine and cytokines and expressing co-stimulatory molecules that modulate the maturation state of dendritic cells. Studies in a murine contact hypersensitivity model show that mast cell-derived tumor necrosis factor (TNF)α is responsible for efficient DC entry to draining lymph nodes [29]. Other in vitro studies have confirmed histamine and IL-4, in particular, alter DCs ability to prime polarized Th responses [30–33]. Although we can find no differences in the maturation state of DCs as defined by expression of B7 or MHC Class II molecules, the antigen presentation capacity of W/Wv-derived splenic DCs measured ex vivo after immunization is sub-optimal (A. Christy, unpublished observation). Thus, there is evidence that mast cells are affecting some aspects of DC function. There is marked lack of the normal lymph node hypertrophy associated with T-cell activation that could reflect marginal DC migration post-activation [21].
Direct Mast Cell Action on T Cell Priming?
Mast cells are also found in secondary lymphoid organs where they have the potential to directly alter T-cell function. How mast cells are activated in these sites is less clear; however, cytokines produced by activated T cells or DCs may be involved. Cytokines expressed by mast cells that could polarize T helper cells include IL-4 (Th2), IL-12 (Th1), transforming growth factor (TGF)β (Th17 and T regulatory cell), and IL-6 (Th17). Both Th1 and Th17 cells are the important effector cells in this EAE and MS, while T regulatory cells offer some protective effects. Because mast cells are major producers of TGFβ and IL-6 (B. Sayed, unpublished observation), it is tempting to speculate that they have a role in regulating the ratio of Th17 effector to T regulatory cell ratio. We observe that the MOG-specific T-cell response in the lymph node and spleen as measured by γIFN production is slightly delayed and decreased in W/Wv mice as are early activation markers, CD44hi, and CD11ahi [21]. However, we discern no alteration in the numbers of Th17 or T regulatory cells in W/Wv mice post-EAE induction.
Mast Cells and Trafficking?
The trafficking and entry of the autoreactive effector T cells into the CNS is a critical step in frank disease development, and this is compromised in W/Wv mice [21]. The consequences of suboptimal cell trafficking in these mice include massive reductions in the inflammatory response and reduced myelin damage.
These events require what is referred to as a “breach” in the BBB. That is, the local endothelium, which is characterized by extremely tight junctions between cells, becomes permeable. This extent of this permeability correlates with disease severity [34]. There must also be an alteration in adhesion molecule expression on the endothelium providing cells with signals designating inflammatory sites, in this case the CNS. Again, mast cells are well equipped to influence these events. Histamine and serotonin are well-known vasoactive agents that promote vascular leakiness. Mast cell cytokines including TNFα induce expression of vascular adhesion molecules such as vascular cell adhesion molecule [35]. Whether vascular permeability and/or local endothelium changes are direct results of mast cell influence or whether mast cells enhance inflammation thereby indirectly alter trafficking is unclear. Another mast cell product of interest is the leukotriene, LTB4. Acting through BLT1 receptors on both CD4+ and CD8+ T cells, mast cell-derived LBT4 is essential for T-cell migration to inflammatory sites in a model of allergic asthma [36–39].
It is unclear which signals are important in initially regulating entry of autoreactive T cells and other inflammatory cells into the CNS. A commonly accepted paradigm, based on studies in infections or allergic airway disease for example, presumes that a pathogen or allergen elicits a local inflammatory reaction which then targets the trafficking of effector cells to involved tissue sites. Innate immune cells are activated, leading to cytokine-induced changes in the regional endothelium, chemokine expression that attracts specific cell populations and localized vascular permeability. However, in EAE (and probably MS as well), the site of antigen entry is not the CNS but rather a dorsal subcutaneous site. It is possible that all activated T cells irrespective of their antigen specificity circulate into tissues at some low frequency and that only the encounter of autoreactive T cells with myelin antigen in the CNS tissues elicits T-cell reactivation. This reactivation starts the inflammatory cascade (perhaps in part through mast cell expression of relevant mediators) that directs specific cell migration and renders the local endothelium generally more permeable to the entry of additional T cells and other inflammatory cells. There is evidence to support this possibility [40].
Mast Cells and T Regulatory Cells?
The balance between autoreactive T effector cells and T regulatory cell function is clearly an extremely important determinant in the autoimmune disease development [41]. It is believed that T regulatory cells can restore homeostasis and counteract the autoimmune response, particularly early in disease. Many studies have confirmed that Foxp3+ T regulatory cells can suppress EAE [42]. In animal models of type 1 diabetes, disease onset is much more rapid when T regulatory cells are depleted [43]. T regulatory cells accumulate in the pancreatic islets of NOD mice but do not appear to be sufficient to prevent disease [44]. Given their influence on CD4+ and CD8+ T-cell responses and impressive production of TNFα and TGFβ, it is possible that mast cells also affect T regulatory cell development and function and perhaps contribute to a balance between effector and regulatory cell populations. TNFα modulates the numbers of CD4+ CD25+ T regulatory cells in the NOD model as well as human autoimmune disease [45, 46]; TNFα treatment during neonatal life reduces the numbers of T regulatory cells, and administration of anti-TNFα antibodies can reverse this process [45]. TGFβ is a cytokine that drives the development of T regulatory cells from naïve CD4+ T cells in the lymph nodes [47, 48]. Even a brief exposure to TGFβ can block diabetes disease progression [49].
There is evidence for mast cell-dependent T regulatory cell suppression in an allogeneic skin graft model [50]. In this scenario, mast cells are recruited to the site of the foreign skin transplant, an observation that correlates with lack of transplant rejection and increases in the expression of several transcripts that are expressed by mast cells including IL-10, FcɛRI, CCL2, and CCL12. Furthermore, mast cell-deficient mice cannot be rendered tolerant to the allograft. Mast cells could also suppress T-cell activation through expression of PDL1, a molecule that is thought to limit expansion of autoreactive CD4+ and CD8+ T cells. PDL1 is constitutively expressed on mast cells (as well as other cell types), and its expression is greatly increased upon activation ([13] and Sayed and Brown, unpublished observation). Blockage of PDL1 and its receptor PD1 accelerates disease in NOD mice [51]. This acceleration is dependent on events that occur in the pancreatic lymph node early in the disease. It is thought to be the result of the ability of PDL1 to limit expansion of autoreactive CD4+ and CD8+ T cells.
Exquisitely sensitive to tissue environment and capable of rapidly responding to insult with a wide variety of preformed factors, inducible cytokines, and specific chemokines, mast cells are poised to influence a wide variety of immune responses. Despite the emerging interest in mast cells by the traditional immunology community and the numerous studies of their interaction with cells of the adaptive immune system, there is still much to be learned about these versatile cells.
References
Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–86.
Galli SJ, Maurer M, Lantz CS. Mast cells as sentinels of innate immunity. Curr Opin Immunol. 1999;11:53–9.
Galli SJ, Lantz CS. Allergy. Philadelphia: Lippincott-Raven; 1999. p. 1137–84.
Feng BS, He SH, Zheng PY, Wu L, Yang PC. Mast cells play a crucial role in Staphylococcus aureus peptidoglycan-induced diarrhea. Am J Pathol. 2007;171:537–47.
Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol. 2004;4:787–99.
Marshall JS, McCurdy JD, Olynych T. Toll-like receptor-mediated activation of mast cells: implications for allergic disease? Int Arch Allergy Immunol. 2003;132:87–97.
Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature 1996;381:75–7.
Malaviya R, Ikeda T, Ross E, Abraham S. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 1996;381:77–80.
Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. 2005;6:135–42.
Sayed BA, Brown MA. Mast cells as modulators of T-cell responses. Immunol Rev. 2007;217:53–64.
Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol. 2008;26:705–39.
Gregory GD, Raju SS, Winandy S, Brown MA. Mast cell IL-4 expression is regulated by Ikaros and influences encephalitogenic Th1 responses in EAE. J Clin Invest. 2006;116:1327–36.
Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J Immunol. 2006;176:238–48.
Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835–48.
McDevitt H. Closing in on type 1 diabetes. N Engl J Med. 2001;345:1060–1.
Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, et al. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. J Exp Med. 1985;162:1025–43.
Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;26:565–71.
Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21.
Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 1996;85:299–302.
Stinissen P, Raus J, Zhang J. Autoimmune pathogenesis of multiple sclerosis: role of autoreactive T lymphocytes and new immunotherapeutic strategies. Crit Rev Immunol. 1997;17:33–75.
Gregory GD, Robbie-Ryan M, Secor VH, Sabatino JJ Jr, Brown MA. Mast cells are required for optimal autoreactive T cell responses in a murine model of multiple sclerosis. Eur J Immunol. 2005;35:3478–86.
Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191:813–22.
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8.
Ibrahim MZM, Reder AT, Lawand R, Takash W, Sallouh-Khatib S. The mast cells of the multiple sclerosis brain. J Neuroimmunol. 1996;70:131–8.
Johnson D, Seeldrayers PA, Weiner HL. The role of mast cells in demyelination. 1. Myelin proteins are degraded by mast cell proteases and myelin basic protein and P2 can stimulate mast cell degranulation. Brain Res. 1988;44:195–8.
Tanzola MB, Robbie-Ryan M, Gutekunst CA, Brown MA. Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J Immunol. 2003;171:4385–91.
Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801.
Dietsch GN, Hinrichs DJ. Mast cell proteases liberate stable encephalitogenic fragments from intact myelin. Cell Immunol. 1991;135:541–8.
Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cell-associated TNF promotes dendritic cell migration. J Immunol. 2006;176:4102–12.
Caron G, Delneste Y, Roelandts E, Duez C, Bonnefoy JY, Pestel J, Jeannin P. Histamine polarizes human dendritic cells into Th2 cell-promoting effector dendritic cells. J Immunol. 2001;167:3682–6.
Mazzoni A, Siraganian RP, Leifer CA, Segal DM. Dendritic cell modulation by mast cells controls the Th1/Th2 balance in responding T cells. J Immunol. 2006;177:3577–81.
Mazzoni A, Young HA, Spitzer JH, Visintin A, Segal DM. Histamine regulates cytokine production in maturing dendritic cells, resulting in altered T cell polarization. J Clin Invest. 2001;108:1865–73.
Yao Y, Li W, Kaplan MH, Chang CH. Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by dendritic cells. J Exp Med. 2005;201:1899–903.
Fabis MJ, Scott GS, Kean RB, Koprowski H, Hooper DC. Loss of blood–brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc Natl Acad Sci USA. 2007;104:5656–61.
Lee CW, Lin CC, Luo SF, Lee HC, Lee IT, Aird WC, Hwang TL, Yang CM. Tumor necrosis factor-alpha enhances neutrophil adhesiveness: induction of vascular cell adhesion molecule-1 via activation of Akt and CaM kinase II and modifications of histone acetyltransferase and histone deacetylase 4 in human tracheal smooth muscle cells. Mol Pharmacol. 2008;73:1454–64.
Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–90.
Taube C, Miyahara N, Ott V, Swanson B, Takeda K, Loader J, et al. The leukotriene B4 receptor (BLT1) is required for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J Immunol. 2006;176:3157–64.
Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol. 2003;4:974–81.
Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol. 2003;4:965–73.
Smorodchenko A, Wuerfel J, Pohl EE, Vogt J, Tysiak E, Glumm R, et al. CNS-irrelevant T-cells enter the brain, cause blood–brain barrier disruption but no glial pathology. Eur J Neurosci. 2007;26:1387–98.
Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–44.
O’Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol. 2008;193:1–11.
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000;12:431–40.
Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92.
Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci USA. 2002;99:12287–92.
Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–85.
Fu S, Zhang N, Yopp AC, Chen D, Mao M, Zhang H, et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25-precursors. Am J Transplant. 2004;4:1614–27.
Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25-T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–53.
Peng Y, Laouar Y, Li MO, Green EA, Flavell RA. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci USA. 2004;101:4572–7.
Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 2006;442:997–1002.
Guleria I, Gubbels Bupp M, Dada S, Fife B, Tang Q, Ansari MJ, et al. Mechanisms of PDL1-mediated regulation of autoimmune diabetes. Clin Immunol. 2007;125:16–25.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brown, M.A., Sayed, B.A. & Christy, A. Mast Cells and the Adaptive Immune Response. J Clin Immunol 28, 671–676 (2008). https://doi.org/10.1007/s10875-008-9247-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-008-9247-7