
Intravenous immunoglobulin (IVIg) infusions at 3–4 week intervals are currently standard therapy in the United States for patients with primary immune deficiency diseases (PIDD). To evaluate alternative modes of immunoglobulin administration we have designed an open-label study to investigate the efficacy and safety of a subcutaneously administered immunoglobulin preparation (16% IgG) in patients with PIDD. After their final IVIg infusion, 65 patients entered a 3-month, wash-in/wash-out phase, designed to bring patients to steady-state with subcutaneously administered immunoglobulin. This was followed by 12 months of weekly SCIg infusions, at a dose determined in a pharmacokinetic substudy to provide noninferior intravascular exposure. This resulted in a mean weekly dose of 158 mg/kg, calculated to equal 137% of the previous intravenous dose. Two patients (4%) each reported 1 serious bacterial infection (pneumonia), an annual rate of 0.04 per patient-year. There were 4.43 infections of any type per patient-year. Mean trough serum IgG levels increased from 786 to 1040 mg/dL during the study, a mean increase of 39%. The most frequent treatment-related adverse event was infusion-site reaction, reported by 91% of patients; this was predominantly mild or moderate, and the incidence decreased over time. No treatment-related serious adverse events were reported. We conclude that subcutaneous administration of 16% SCIg is a safe and effective alternative to IVIg for replacement therapy of PIDD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Immunoglobulin replacement has been standard therapy for patients with primary immune deficiency diseases (PIDD) since it was introduced more than 50 years ago by Bruton and colleagues to treat a patient with agammaglobulinemia (1). Long-term treatment, using human immunoglobulin derived from donated plasma, reduces the frequency and severity of infections by maintaining serum immunoglobulin G (IgG) concentrations closer to physiological levels in the normal population. Although Bruton initially administered immunoglobulin subcutaneously, intravenous administration became the preferred mode of administration in the 1980s. Intravenously administered immunoglobulin (IVIg) allowed infusion of higher doses over a relatively short time, and has remained the standard route of administration in much of the world. However, IVIg therapy is not ideal for all patients and may be difficult for those with poor venous access or those experiencing recurrent systemic reactions. Reactions to IVIg therapy that persist beyond the first infusion are usually mild to moderate and include myalgia, fever, chills, headache, lower back pain, nausea, and vomiting (2–4). The incidence of these reactions has decreased substantially with the development of newer IVIg preparations, such that they are now estimated to affect only approximately 5% of patients (5). Nevertheless, some studies have reported substantially higher rates of headache, up to 61% (6, 7). Although the incidence of systemic reactions can be reduced by decreasing the rate of infusion, many patients require premedication, including hydrocortisone, prior to their IVIg infusion. In a recent survey by the Immune Deficiency Foundation, 45% of the responding patients reported that they regularly premedicate before receiving IVIg infusions (8).
Several clinical trials of subcutaneously administered infusions of immunoglobulin, formulated for intramuscular use, provided high serum trough levels of IgG and comparable protection from infection, while adverse events were reduced when compared to IVIg (2, 9–14). In these trials, subcutaneous IgG was administered every 3 to 14 days, as compared to every 21 to 28 days for IVIg, and provided more consistent serum IgG trough concentrations compared to the initial high peak followed by low trough levels associated with IVIg therapy.
Studies in Sweden, where subcutaneous infusions of immunoglobulin have become standard, and recently in North America have also demonstrated significant improvement in quality of life and treatment satisfaction as reported by PIDD patients due to the increased independence and scheduling flexibility associated with home-based, self-administered therapy (12, 13, 15, 16). In these studies, patients reported that self-administration of subcutaneous IgG was easy to learn. Therefore, in addition to patients with poor venous access and those who experience intolerable systemic adverse events with IVIg therapy, appropriate candidates for subcutaneous therapy may include motivated, independent patients, and/or patients who live far from an infusion center or have time constraints during business hours of infusion centers. Since no approved immunoglobulin preparation for subcutaneous use is presently available on the US market, this route of administration has been available for only a fraction of patients who may otherwise have opted for subcutaneous IgG therapy.
Vivaglobin® (ZLB Behring: Marburg, Germany and Bern, Switzerland; SCIg), a polyvalent unmodified human immunoglobulin, has been licensed for subcutaneous administration in several European countries and in the US by the Food and Drug Administration (FDA). The present study was designed to evaluate the efficacy, safety, and tolerability of subcutaneously administered SCIg in children and adults with PIDD.
METHODS
Patient Selection
Male and non-pregnant female patients with predominant antibody deficiencies (common variable immune deficiency and X-linked agammaglobulinemia) were eligible for study entry. Patients had to be on IVIg therapy for at least 4 months, over 2 years of age and have a bodyweight of 10 kg or more. Exclusion criteria included evidence of hepatitis (A, B or C) or HIV infection at screening, bleeding disorders, and treatment with immunosuppressive drugs. Individuals with a history of alcohol or drug abuse, breastfeeding women, and women of childbearing age not using adequate contraception were also excluded from the study.
Study Design
The clinical trial was designed as an open-label, prospective, multicenter study, in accordance with the International Conference on Harmonisation (ICH), Good Clinical Practice (GCP) guidelines and the Declaration of Helsinki. The study received Institutional Review Board approval, and prior to enrolling into the study, all patients provided their written informed consent.
The study included three phases: baseline (3–4 weeks), wash-in/wash-out (12 weeks), and efficacy (52 weeks). During the baseline phase, each patient received his/her usual IVIg treatment; vital signs, baseline biochemical and viral tests were performed, and serum IgG trough levels were measured. One week after receiving the last IVIg dose, each patient started weekly subcutaneous infusions of SCIg (Vivaglobin® 16% IgG; ZLB Behring: Marburg, Germany and Bern, Switzerland) for at least 3 months (wash-in/wash-out phase). Prior to starting this phase, all patients or their caregivers were instructed on how to administer SCIg therapy at home. The first two subcutaneous infusions were done under the supervision of trained nurses. The product was administered at 1 or multiple injection sites using an infusion pump, with rates limited to ≤20 ml/h at any 1 injection site, and a total volume of ≤15 ml per injection site.
The dosage of SCIg used in this study was based on the results of a pharmacokinetic study carried out in a subset of 24 patients, to determine the area under the curve for both IVIg and SCIg. The results of this investigation determined that to achieve area under the curve for serum IgG over time comparable to previous IVIg therapy, the total monthly SCIg dose must be 137% of the previous IVIg dose. For those patients participating in the pharmacokinetic substudy, the SCIg doses were individually adjusted based on each subject's IgG levels. All other patients entering the efficacy phase received a doe adjustment of 137% of their weekly-equivalent IVIg dose, which resulted in a mean weekly dose of 158 mg/kg (range 155–165 mg/kg).
Efficacy Assessments
The primary efficacy variable was the number of clinically documented serious bacterial infections (SBIs) occurring during the 12-month efficacy period. SBIs were defined as bacterial pneumonia, meningitis, sepsis, osteomyelitis, or visceral abscess. These were diagnosed by a practicing physician (preferably the study investigator) according to standard medical procedures (physical examination, laboratory tests, bacterial cultures, imaging).
Secondary efficacy variables included episodes of infection (protozoa, bacteria, fungi, or viruses) other than those identified as SBIs. The number and duration of febrile episodes (>38.5°C) were also recorded. The use of antibiotics during the efficacy phase was documented, including the dose and the number of treatment days. The number of days off school or work due to infections and the number of days in hospital due to infections were recorded by each patient in a diary. Trough levels of serum IgG were measured on serum samples taken immediately prior to the efficacy-phase subcutaneous infusions on days 1, 17, 29, 41, and 53.
Adverse events (AEs), laboratory-safety variables, and blood-borne virus screening were documented throughout the study. AEs were classed as treatment-related if they appeared after the onset of SCIg therapy. Vital signs (e.g., pulse and blood pressure) and infusion-site reactions were monitored before infusion, during the procedure, and 30 min after completion of the infusions. These measurements were performed during every IVIg infusion and every fourth SCIg infusion.
Statistical Analysis
All efficacy variables were analyzed according to the protocol; only those patients who completed the 12-months efficacy phase were evaluated. The primary objective of the study was to demonstrate that the number of SBIs/patient-year was <1. Poisson regression was used to calculate the upper 99% confidence limit (CL) of the annual SBI rate. Sensitivity analyses were also conducted to consider data collected from the 14 discontinued patients and extrapolate the number of SBIs per subject, thus avoiding introducing any bias into the study findings.
The safety analyses were based on a modified intention-to-treat (ITT) population: all patients that had received at least one SCIg infusion were included. Safety variables were assessed using appropriate descriptive statistical methods.
RESULTS
Patient Disposition
Patient demographics and disposition are described in Table I. A total of 68 patients were enrolled into the study and entered the baseline IVIg phase (ITT population). Since three patients withdrew before receiving any study medication, 65 started the 3-month, wash-in/wash-out phase (modified ITT population). As 14 patients withdrew from the study after starting the efficacy phase, a total of 51 patients completed the study according to the protocol (per-protocol population). The most common reasons for withdrawal were adverse events and withdrawal of consent (six patients each); one patient was lost to follow-up and there was one protocol violation. The flow of patients through the study is provided in Fig. 1.

Flow of patients through the study.
Serum Trough IgG Levels
Trough levels of serum IgG were higher during the efficacy phase of the study than at baseline. The mean pre-infusion level increased from 786 mg/dL at baseline to 1,040 mg/dL during SCIg treatment, representing a 39% increase. As shown in Fig. 2, trough levels remained relatively stable throughout the wash-in/wash-out and efficacy phases of the study.

Mean serum IgG trough concentrations during the study.
Infections
During the 12-month efficacy phase, 2 SBIs were reported, resulting in an annual rate of 0.04 episodes per patient-year (upper 99% CL: 0.14). Both SBIs were pneumonias, and two patients (4%) were affected. Sensitivity analyses produced values ranging between 0.04 and 0.07 SBIs per patient-year.
A total of 49 patients (96%) reported infections of any type during the efficacy phase. This group includes 11 patients reporting chronic infections (sinusitis, bronchitis, conjunctivitis, dental abscess) with durations longer than 220 days. The annual rate of infections was 4.43 per patient-year, and the number of days with infection was 118.0 per patient-year. There was an apparent trend towards overall infection rate reductions with increasing age (Table II). The most frequently reported infections were sinusitis (in 57% of patients), upper respiratory infection (51%), bronchitis (25%), rhinitis (20%), and conjunctivitis (20%) (Table II). Sinusitis was the longest lasting infection (mean duration of 53 days, suggesting that sinusitis is a chronic problem in this patient population), followed by upper respiratory infection (22 days).
Nine febrile episodes were observed during the efficacy phase in 6 patients (11.8%). The cumulative duration of these episodes was 12 days, giving an annualized rate of 0.23 days per patient-year. Forty-seven patients (92.2%) reported using antibiotics, for a total of 6,240 days. The annual rate of antibiotic use was 120.2 days per patient-year.
Thirty-two patients (62.7%) missed a total of 192 days of school or work due to infections during the efficacy phase, resulting in an overall rate of 3.70 days per patient-year. Over the same period, four patients (7.8%) were hospitalized due to infection (including the two patients with pneumonia), for a total of 12 days or 0.23 hospital days per patient-year.
Adverse Events
Out of a total of 3,656 infusions, 2,584 treatment-emergent AEs were reported (0.71 per infusion), and 1,901 were considered by the investigator to be treatment related (0.52 per infusion). There was no relationship between the incidence or rate of treatment-emergent AEs and increasing the dose of SCIg.
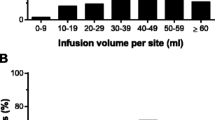
Details of treatment-related adverse events are given in Table III. The most frequent type of treatment-emergent AE, infusion-site reaction, was observed at least once in 60 patients (91%). The large majority (96%) of these reactions were of mild or moderate intensity; most were of short duration (1 or 2 days); and most required no treatment. As shown in Fig. 3, the incidence of infusion-site reactions decreased over time. While 85% of patients (55/65) reported infusion-site reactions after the first SCIg infusion, this had declined to 41% (22/54) after the 33rd infusion and remained relatively stable thereafter. Three subjects discontinued treatment due to infusion-site reactions—the withdrawals were on days 1, 22, and 78 after SCIg therapy was started.

Incidence of infusion-site adverse events over time.
Another treatment-emergent adverse event reported in this study was headache. A total of 31 subjects (48%) reported a total of 159 episodes of headache during the one year efficacy period. Of these 31 subjects, 11 reported mild headache, 9 moderate, and 11 severe. The mean duration of headache was 2.7 days, and mean time to onset of headache in most subjects was 2 days after infusion. There were 59 episodes of headache, which were considered treatment-related and there were a total of 3,656 infusions. Thus, the rate of infusion-related headaches was 1.6%.
Nine patients (14%) reported a total of 10 serious adverse events (surgery [1], atrial septal defect [1], postural hypotension [1], gastroenteritis [1], intestinal obstruction [1], other gastrointestinal disorder [1], depression [1], epistaxis [1], and pneumonia [2]), none of which were related to study treatment. No deaths were reported during the study. No hematological or other laboratory parameter showed any notable changes during the study, and no product-related, virus-safety issues were apparent.
DISCUSSION
The results of this study demonstrate the efficacy, safety, and tolerability of self-administered subcutaneous immunoglobulin therapy. The annualized rates of both serious bacterial infection (0.04/patient) and overall infection (4.43/patient) are low. It is difficult to compare these results with other studies owing to differences in dosing, patient selection, infection reporting and other aspects of methodology. Nevertheless, the results appear similar to those obtained with IVIg therapy: two recent studies of IVIg in PIDD patients have shown SBI rates of 0.1 (17) and 0.061 (18) per patient-year, respectively, and an overall infection rate of 3.9 per patient-year (19). Furthermore, in a parallel study of SCIg conducted in Europe and Brazil (reported in the same issue of the Journal), the same immunoglobulin preparation as used in the present study was found to produce almost identical rates of overall infections and serious infections (20). The primary difference between these two studies is the weekly dose: in the European/Brazilian study, the SCIg dose was calculated to be equivalent to 100% of each patient's previous IVIg dose, while in the North American study this was 137%. The results from the European/Brazilian study demonstrate that administering a SCIg dose equal to the standard IVIg dose (approximately 100 mg/kg per week) results in an IgG trough level (920 mg/dL) that provides adequate protection to these patients while not being associated with additional SBIs. Conversely, the increased IgG trough levels (mean 1040 mg/dL) observed in the current study do not appear to confer any additional benefits. This reflects the findings of an earlier study where no dose-related differences in infection rates were found between IVIg administered monthly at doses of 200, 400, or 600 mg/kg (21).
The overall infection rate observed during the present study necessitated treatment with antibiotics in about 1 day of 3. However, most reported infections were minor and did not result in a loss of time from school or work: each patient missed on average fewer than 4 days of school or work per year and only four patients had to be hospitalized due to infections for a total of 12 days, which is less than a quarter of a day per patient per year.
The present study demonstrates that SCIg therapy is well tolerated, with no apparent safety concerns. Although 97% of patients experienced treatment-related AEs, this was primarily attributable to a high rate of infusion-site reactions including local swelling, soreness, redness, and induration, which affected 92% of patients and were almost all of mild or moderate intensity. This finding is consistent with previous studies, which have reported similar levels of local tissue reactions following subcutaneous administration of immunoglobulin preparations (10, 14, 20). It is interesting to note that in the European/Brazilian study using the same SCIg preparation, the Swedish subjects who had been receiving long-term SCIg therapy prior to study entry reported virtually no infusion-site reactions (20).
Approximately one in five patients withdrew prematurely from the study. Apart from consent withdrawal by six patients (which would not be encountered in clinical practice), the most common reason for withdrawal was AEs. However, only three subjects discontinued treatment due to infusion-site reactions. The rate of withdrawal due to AEs can be minimized by ensuring optimal patient education, so that the product is administered with the correct technique and the patient recognizes that some local reaction is to be expected. Successful SCIg therapy is also dependent on convincing the patient that maximum adherence to the infusion schedule is essential to treatment efficacy.
The efficacy data collected during this study are similar to those reported from previous trials using other subcutaneously administered immunoglobulin preparations (2, 10, 22). A clinical trial by Gaspar et al. involving children with PIDD reported no serious or life-threatening infections and no infections requiring hospitalization during an observation period of between 6 months and 3.5 years (22). Subcutaneous administration was well tolerated, with no systemic or clinically significant AEs observed (2, 10, 22).
In a direct comparison of SCIg versus IVIg replacement therapy, no significant differences between the number of infections were reported, with overall infection rates of 3.82 and 4.12, respectively (10). The subcutaneous administration of immunoglobulin has a number of potential benefits over intravenous infusion that may appeal to many PIDD patients. These include elimination of venous access; improved safety; smoother, more consistent, and stable serum IgG levels; and the flexibility provided by self-infusion at home rather than in an infusion center (13).
In addition to the benefits of decreased systemic AEs associated with SCIg administration, the potential risk of life-threatening anaphylactic shock may be reduced or even eliminated. Patients with antibodies to immunoglo-bulin A (IgA) have been identified as being potentially at risk of anaphylactic shock during IVIg therapy (23). Clinical observations suggest that SCIg can not only be administered safely to these patients, but may even lead to a decrease or elimination of anti-IgA antibodies (24, 25).
One unresolved issue is the optimal dosing of SCIg in comparison to IVIg and the resulting IgG peak and trough levels. A single monthly IVIg dose, usually 400 mg/kg, results in an instant rise in serum IgG level with an average increase of 800–1,000 mg/dL above the trough level. On subsequent days serum IgG concentrations drop relatively quickly due to the equilibration of the infused immunoglobulin between the intravascular and total extracelluar spaces (26). This equilibration phase is followed by a slow decline in serum IgG concentration, which is dependent on the metabolic rate of the patient and the half-life of the preparation. It is generally recommended that the trough IgG level at the time of the next infusion remains at or above 500–600 mg/dL, or 350–450 mg/dL above the baseline (the IgG level prior to starting IVIg) (27, 28). In contrast to IVIg, frequent subcutaneous infusions of IgG will generate a local depot resulting in slow absorption and in a nearly constant serum level of IgG. In this trial the mean “trough” IgG level was 1,040 mg/dL. Thus, in contrast to the high peak and low trough levels observed with monthly infusions of IVIg, weekly infusions of SCIg generate peak and trough levels that differ very little. Because the rate of SBIs was only 0.04 per patient per year, it is unlikely that a high serum IgG peak is required for the success of immunoglobulin prophylaxis. On the other hand, the low trough level of IgG has been linked to the increased susceptibility of PIDD patients to bacterial infections during the week before the monthly IVIg infusions.
Finally, the controversy of calculating comparable doses for SCIg and IVIg needs to be addressed. After consulting the FDA, a dose of SCIg was chosen that led to an area under the curve equal to that achieved by IVIg. This postulate required an average increase of the SCIg dose to 137% of that given intravenously. However, such an increase in the SCIg dose seems to be unnecessary since the rate of serious infection during the North American study was the same as during the European/Brazilian study in spite of the fact that in the European/Brazilian clinical trial the monthly dose of SCIg was kept identical to the IVIg dose.
Most importantly, all participating patients managed without difficulties the task of self infusion at home. Self-infusion of IVIg at home is more difficult and is offered by very few centers in the United States. Quality-of-life studies have shown consistently that self infusion at home is crucial for patient satisfaction (15, 16). Thus, the availability of preparations that can be infused subcutaneously will provide PIDD patients with the option of self-infusion at home without compromising safety and efficacy, and potentially at lower cost (14).
In conclusion, the data from this open, prospective, multicenter study confirm that self-administered, subcutaneous immunoglobulin therapy is a safe and effective alternative to intravenous immunoglobulin therapy for children and adults with PIDD.
Subcutaneous IgG Study Group/Investigators
Arthur Althaus, MD, Louisville, KY; Pedro C Avila, MD, San Francisco, CA; Melvin Berger, MD, PhD, Cleveland, OH; Sudhir Gupta, MD, PhD, MACP, Irvine, CA; Harry Hill, MD, Salt Lake City, UT; Robert W Hostoffer, DO, South Euclid, OH; Lisa J Kobrynski, MD, FRCP, Atlanta, GA; Robyn Levy, MD, Atlanta, GA; Laurie Myers, MD, Durham, SC; Hans D Ochs, MD, Seattle, WA; G Wendell Richmond, MD, Oak Brook, IL; Robert L Roberts, MD, PhD, Los Angeles, CA; Chaim Roifman, MD, FRCP, FCACB, Ontario, Canada; Ralph Shapiro, MD, Plymouth, MN; Suzanne Skoda-Smith, MD, Gainesville, FL; Donald Stark, MD, Vancouver, British Columbia; Mark Stein, MD, North Palm Beach, FL; S Bobo Tanner, MD, Nashville, TN; Peter Vadas, MD, PhD, Toronto, Canada.
Abbreviations
- AEs:
-
adverse events
- CL:
-
confidence limit
- HIV:
-
human immunodeficiency virus
- ITT:
-
intention-to-treat
- IgA:
-
immunoglobulin A
- IgG:
-
immunoglobulin G
- PIDD:
-
primary immune deficiency diseases
- IVIg:
-
intravenously administered immunoglobulin
- SCIg:
-
subcutaneously administered immunoglobulin
- SBIs:
-
serious bacterial infections.
REFERENCES
Bruton OC: Agammaglobulinemia. Pediatrics 9:722–728, 1952.
Björkander J, Chapel H, Spickett G: Comparison of the efficacy and safety of immunoglobulin given subcutaneously versus intravenous immunoglobulin in the prevention of infection in patients with primary antibody deficiency syndromes. Mol Immunol 35:11–12, 1998.
Cunningham-Rundles C, Siegel F, Smithwick E, Lion-Boule A, Cunningham-Rundles S, O’Malley J: Efficacy of intravenous immunoglobulin in primary humoral immunodeficiency disease. Ann Intern Med 101:435–439, 1984.
Lederman H, Roifman C, Lavi S, Gelfand E: Corticosteroids for prevention of adverse reactions to intravenous immune globulin infusions, in hypogammaglobulinemic patients. Am J Med 81:443–446, 1986.
Ratko TA, Burnett DA, Foulke GE, Matuszewski KA, Sacher RA: Recommendations for off-label use of intravenously administered immunoglobulin preparations. University Hospital Consortium Expert Panel for Off-Label Use of Polyvalent Intravenously Administered Immunoglobulin Preparations. JAMA 273:1865–1870, 1995.
Bertorini TE, Nance AM, Horner LH, Greene W, Gelfand MS, Jaster JH: Complications of intravenous gammaglobulin in neuromuscular and other diseases. Muscle Nerve 19:388–391, 1996.
Brannagan TH, 3rd, Nagle KJ, Lange DJ, Rowland LP: Complications of intravenous immune globulin treatment in neurologic disease. Neurology 47:674–677, 1996.
Immune Deficiency Foundation 2003, posting date. Treatment experiences and preferences of patients with primary immune deficiency disease: First national survey. [Online.]
Berger M, Cupps TR, Fauci AS: Immunoglobulin replacement therapy by slow subcutaneous infusion. Ann Intern Med 93:55–56, 1980
Chapel HM, Spickett GP, Ericson D, Engl W, Eibl MM, Bjorkander J: The comparison of the efficacy and safety of intravenous versus subcutaneous immunoglobulin replacement therapy. J Clin Immunol 20:94–100, 2000.
Gardulf A, Hammarstrom L, Smith CI: Home treatment of hypogammaglobulinaemia with subcutaneous gammaglobulin by rapid infusion. Lancet 338:162–166, 1991
Gardulf A, Bjorvell H, Gustafson R, Hammarstrom L, Smith CI: The life situations of patients with primary antibody deficiency untreated or treated with subcutaneous gammaglobulin infusions. Clin Exp Immunol 92:200–204, 1993.
Gardulf A, Hammarstrom L: Subcutaneous administration of immunoglobulins: What are the advantages? Clin Immunother 6:108–116, 1996.
Gardulf A, Andersen V, Bjorkander J, Ericson D, Froland SS, Gustafson R, Hammarstrom L, Jacobsen MB, Jonsson E, Moller G, et al.: Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: Safety and costs. Lancet 345:365–369, 1995.
Gardulf A, Nicolay U, Math D, Asensio O, Bernatowska E, Bock A, Costa-Carvalho BT, Granert C, Haag S, Hernandez D, Kiessling P, Kus J, Matamoros N, Niehues T, Schmidt S, Schulze I, Borte M: Children and adults with primary antibody deficiencies gain quality of life by subcutaneous IgG self-infusions at home. J Allergy Clin Immunol 114:936–942, 2004.
Nicolay U, Kiessling P, Berger M, Gupta S, Yel L, Roifman CM, Gardulf A, Eichmann F, Haag S, Massion C, Ochs HD: Health-related quality of life and treatment satisfaction in north american patients with primary immunedeficiency diseases receiving subcutaneous IgG self-infusions at home. J Clin Immunol 26:65–72, 2006.
Ochs HD, Pinciaro PJ: Octagam 5%, an intravenous IgG product, is efficacious and well tolerated in subjects with primary immunodeficiency diseases. J Clin Immunol 24:309–314, 2004.
Berger M: Subcutaneous immunoglobulin replacement in primary immunodeficiencies. Clin Immunol 112:1–7, 2004.
Eijkhout HW, van Der Meer JW, Kallenberg CG, Weening RS, van Dissel JT, Sanders LA, Strengers PF, Nienhuis H, Schellekens PT: The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann Intern Med 135:165–174, 2001.
Gardulf A, Nicolay U, Asensio O, Bernatowska E, et al.: Rapid subcutaneous IgG replacement therapy is effective and safe in children and adults with primary immunodeficiencies—a prospective, multi-national study. J Clin Immunol In press, 2006.
Pruzanski W, Sussman G, Dorian W, Van T, Ibanez D, Redelmeier D: Relationship of the dose of intravenous gammaglobulin to the prevention of infections in adults with common variable immunodeficiency. Inflammation 20:353–359, 1996.
Gaspar J, Gerritsen B, Jones A: Immunoglobulin replacement treatment by rapid subcutaneous infusion. Arch Dis Child 79:48–51, 1998.
Burks AW, Sampson HA, Buckley RH: Anaphylactic reactions after gamma globulin administration in patients with hypogammaglobulinemia. Detection of IgE antibodies to IgA. N Engl J Med 314:560–564, 1986.
Sundin U, Nava S, Hammarstrom L: Induction of unresponsiveness against IgA in IgA-deficient patients on subcutaneous immunoglo-bulin infusion therapy. Clin Exp Immunol 112:341–346, 1998.
Eijkhout HW, van den Broek PJ, van der Meer JW: Substitution therapy in immunodeficient patients with anti-IgA antibodies or severe adverse reactions to previous immunoglobulin therapy. Neth J Med 61:213–217, 2003.
Berger M, Pinciaro PJ: Safety, efficacy, and pharmacokinetics of Flebogamma 5% [immune globulin intravenous (human)] for replacement therapy in primary immunodeficiency diseases. J Clin Immunol 24:389–396, 2004.
Ochs HD, Fischer SH, Wedgwood RJ, Wara DW, Cowan MJ, Ammann AJ, Saxon A, Budinger MD, Allred RU, Rousell RH: Comparison of high-dose and low-dose intravenous immunoglobulin therapy in patients with primary immunodeficiency diseases. Am J Med 76:78–82, 1984.
Stiehm ER: Human intravenous immunoglobulin in primary and secondary antibody deficiencies. Pediatr Infect Dis J 16:696–707, 1997.
ACKNOWLEDGMENTS
The clinical investigation was sponsored by ZLB Behring, Marburg, Germany. Dr Kiessling is employed by ZLB Behring; Dr Berger has received research support and consulting fees from ZLB Behring. We thank all of the personnel involved in this investigation.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Ochs, H.D., Gupta, S., Kiessling, P. et al. Safety and Efficacy of Self-Administered Subcutaneous Immunoglobulin in Patients with Primary Immunodeficiency Diseases. J Clin Immunol 26, 265–273 (2006). https://doi.org/10.1007/s10875-006-9021-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-006-9021-7