
Abstract
Thiophene-2,5-dicarbonyl dichloride was treated with pyrazole and 3-methylpyrazole (3-MePz) in the presence of trimethylamine. Under normal conditions at room temperature, thiophene-2,5-diylbis((1H-pyrazol-1-yl)methanone) (1) and thiophene-2,5-diylbis((3-methyl-1H-pyrazol-1-yl)methanone) (2) were afforded, respectively. Structure of compounds was deduced from the characteristic 1H and 13C-NMR data set. The structure of compound 2 was also confirmed by X-ray diffraction. Selected parameters of compound 1 and structural parameters of compound 2 were calculated by DFT using B3LYP/G-311 level of theory. The calculated data (bond lengths and angles) were found in close agreement with the experimental data. The compound 2 in solid state has planar structure, the free rotation of 3-methylpyrazolyl group has probably been restricted by some sort of intermolecular interactions. In contrast the gas phase optimized structure shows that 3-MePz rings stabilize themselves at maximum distance from each other.
Graphical Abstract
Pyrazolyl and 3-methylpyrazolyl substituted compounds derived from thiophene-2,5-dicarbonyl dichloride were obtained. The X-ray structure of 3-methylpyrazolyl substituted derivative was established, the solid state data was also calculated by DFT. The experimental structure shows high degree of planarity while optimized structure allows free rotation of the substituents.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is enormous interest in the syntheses of thiophene derivatives [1] because of their key function in fulfilling the everyday life’s needs particularly in medicinal chemistry [2, 3]. They have shown anti-inflammatory [2, 4–7] anti-depressive, analgesic [5, 8], anti-allergic [9], anti-fungal, anti-bacterial [2] and anti-cancer [10] activities. They have been reported to be estrogen-antagonists [11] and are efficient carbonic anhydrase inhibitors [12]. The thiophene derived compounds are attractive synthones for designing interesting materials [13] i.e. low band gap semi-conductors, high potential semi-conductors [14, 15], photovoltaic materials [16–18], luminescent materials [19, 20] and high performance polymer solar cells [21–23]. Most of the compounds containing thiophene as parent ring are derived through substitution at 2 and 5 positions. Such substitution reactions have afforded compounds being used as organic solar cells [24], chemosensors [25], have excellent electronic properties [26] and highly selective anion sensing potency [27]. The 2,5-linked pyridine–thiophene oligomers have been reported to exhibit strong absorption and emission properties. These derivatives show excessive tolerance against oxidation and readily get reduced [28]. Thiophene derivatives are accessible through various synthetic ways [29, 30].
Taking thiophene-2,5-dicarbonyl dichloride as starting precursor, the Cl− function was substituted by pyrazole derivatives. The synthetic route is straightforward and afforded no side reactions. New compounds were characterized with the help of 1H and 13C NMR spectroscopy and structure of compound 2 was confirmed by X-ray diffraction. Structural features of compound 2 were calculated by DFT. Solid state (experimental) and gaseous phase (theoretical) structures are compared and discussed.
Experimental
General
All manipulations were performed in Schlenk-type glassware. Carefully dried solvents and oven-dried glassware were used throughout. The thiophene-2,5-dicarbonyldichloride, pyrazole, 3-methylpyrazole (3-MePz) and trimethylamine were commercial products and were used without further purification. NMR measurements in CDCl3 (concentration ca. 10–15 %) with samples in 5 mm tubes at 23 ± 1 °C: Varian Inova 300 MHz spectrometers for 1H, and 13C NMR; chemical shifts are given relative to Me4Si [δ 1H (CHCl3) = 7.23; δ 13C (CDCl3) = 77.0; chemical shifts are given to ±0.1 for both 1H and 13C].
All quantum-chemical calculations were carried out using the Gaussian 03 program package. Optimized geometries at the B3LYP/6-311+G level of theory were found to be minima by the absence of imaginary frequencies [31–36].
Synthesis of Thiophene-2,5-diylbis((1H-pyrazol-1-yl)methanone) 1 and Analogous Compound 2
For syntheses of compounds 1 and 2 the standard literature procedure was used [37, 38]. The thiophene-2,5-dicarbonyldichloride (0.93 g, 4.5 mM) in toluene (20 mL) was taken up in a Schlenk tube and the solution was kept on stirring. Triethylmaine in twofold excess (1.5 mL, 9.8 mM) was added followed by slow addition of pyrazole (0.612 g, 9 mM). The desired product was obtained as solid (precipitates). The reaction was allowed to continue overnight to ensure maximum formation of products. The solid was separated from the mother liquor, dissolved in chloroform and washed several times with an excess amount of water. The solvents and other readily volatile materials were evaporated under reduced pressure. Compound 1 was obtained as yellow amorphous powder and its solution state structure was deduced from the characteristic NMR data set. For the preparation of compound 2 the same procedure was adopted. Crystals of compound 2 were obtained in CHCl3, the quality was good enough which allowed for confirmation of the solid state structure.
Thiophene-2,5-diylbis((1H-pyrazol-1-yl)methanone), 1: yield: 72 %; 1H-NMR (300 MHz, CDCl3): δ = 8.42 (s, 2H, thiophene), 7.89 (d, 4H, CH) 6.58 (q, 2H, CH) ppm; 13C-NMR (75.8 MHz, CDCl3): δ = 110.3, 129.8, 136.8, 141.3, 144.6, 158.9 (C=O) ppm.
Thiophene-2,5-diylbis((3-methyl-1H-pyrazol-1-yl)methanone), 2: yield: 77 %; 1H-NMR (300 MHz, C6D6): δ = 8.29 (s, 2H, thiophene), 8.01 (d, 2H, CH, 2.80 Hz), 5.65 (d, 2H, CH, 2.80), 1.99 (s, 6H, Me) ppm; 13C-NMR (75.8 MHz): δ = 13.6 (Me), 110.5, 130.1, 136.7, 141.8, 154.0, 158.4 (C=O) ppm.
X-ray Structure Determination
Details pertinent to the crystal structure determinations of compound 2 are listed in Table 1. Crystals of suitable dimensions were selected (in perfluorinated oil [39] at room temperature), and the data collections were carried out at 133(2) K, using a STOE IPDS II system equipped with an Oxford Cryostream low-temperature unit. Structure solutions and refinements were accomplished using SIR97 [40], SHELXL-97 [41] and WinGX [42]. The data have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC 1052874. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre, www.ccdc.cam.ac.uk/getstructures.
DFT Calculations
The geometry of the thiophene-2,5-diylbis((3-methyl-1H-pyrazol-1-yl)methanone) (2) was obtained from X-ray crystallographic data. The molecular structure of 1 (C12H8N4O2S) and 2 (C14H12N4O2S) in ground state was optimized by DFT method including correlation correction using B3LYP/6-311G model of theory. Mullikan’s charges were calculated for both the compounds by using the same model of theory; the data so-obtained are given in Tables 2, 3, 4 and 5 and optimized structure of the compound 2 is given in Fig. 2. All calculations were performed by using Gauss-view molecule visualizer program and GAUSSIAN-03 program [43, 44].
Results and Discussion
Compounds 1 and 2 (Scheme 1) were prepared by adopting the literature procedure as described in “Experimental” section [37, 38]. Both the compounds 1 and 2 were obtained as solid material and were isolated from the mother liquor unambiguously. Compound 1 was obtained as powder and our attempts to get its crystals were unsuccessful.
The synthesized compounds 1 and 2 show 1H-NMR signals at 8.42 and 8.29 ppm which can be assigned to CH of the thiophene ring, respectively. Protons correspond to the pyrazolyl groups were observed in the expected regions. The 13C-NMR data show a characteristic signal at ≈158 ppm which corresponds to CO moiety of the compound. The S–C carbon appears at ≈141 ppm and all other carbons were observed in the expected region. The data obtained for compounds 1 and 2 are well comparable with the data as reported for analogous compound [38].

Syntheses of thiophenene-2,5-dicarbonyl derivatives 1 and 2
Description of Compounds 2
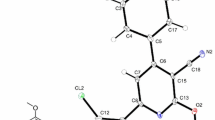
Molecular structure of compound 2 is given in Fig. 1 and the data related to structure determination and refinements are summarized in Table 1. Selected bond lengths and bond angles of compound 2 are represented in Table 2 together with calculated data. The molecule contains a thiophene (Th) as principal ring and two 3-MePz groups attached at 2 and 5 positions. An interesting aspect of the molecule is its planarity and pyrazole and thiophene rings are coplanar. The planarity of molecule is probably because of intermolecular interactions in the crystal packing or some intramolecular interactions of similar nature as reported for structurally analogous compounds [45]. There are some intermolecular interactions between S···S 3.465 Å, C···C 3.395, 3.293 Å and C···N 3.229 Å. These interactions hinder free rotation of the 3-MePz groups around the Th and brings the entire molecule in the same plan (see Table 3 for selected torsion angles). Such type of features either does not exist in 3,5-diMePz substituted molecule or are so weak to bring planarity in the molecule [38] where the 3,5-dimethylpyrazolyl groups are twisted with respect to each other. The geometry around C5 and C10 is trigonal planar with slight distortion. Angles being observed around both the carbon atoms are ∠N1–C10–C1 118.69°, ∠N1–C10–O1 119.43°, ∠O1–C10–C1 121.88° and ∠N3–C5–C4 118.89°, ∠O2–C5–N3 119.26°, ∠O2–C5–C4 121.85°. The narrow angle ≈118° is probably because of the interaction between unsubstituted N atom of the pyrazolyl group and S atom of the thiophene ring.

Molecular structure of compound 2 with partial numbering scheme, ellipsoids are drawn at 50 % probability level. For selected bond lengths and angles see Table 2
The optimized structure of the compound 2 is given in Fig. 2. The calculated data obtained for the compound under discussion are in close agreement with the experimental data (see Table 2). The optimized structure of the molecule shows that one pyrazolyl group is considerably twisted with respect to the principal ring (Table 3).

Optimized structure of compound 2, with partial numbering scheme. 3-methylpyrazolyl groups are randomly arranged in the space in contrast to the actual picture as shown in Fig. 1 above
Mullikan’s charges distribution of compounds 1 and 2 were calculated at the same level of theory, Table 4. The data represented for selected atoms show that considerable negative charge density is accumulated on N1–N4 i.e., −0.343, −0.310, −0.341, −0.364 (compound 2) the same density is slightly different on N2 and N4 of compound 1, which is probably because of Me group present in compound 2. The S atoms of thiophene rings in both the compounds bear an identical positive charge of magnitude 0.372. The calculated data related to Mulikan’s charges on atoms of the compounds predict certain intermolecular interactions in solid state. By comparing the charges of compounds 1 and 2 with our recently published thiourea derivative [46] the electron density is comparatively low on S as well as N atoms.
Some of the electronic properties of the compounds 1 and 2 are summarized in Table 5, which were calculated at the same level of theory (B3LYP/6-311G). The energy gap between HOMO and LUMO is reasonably small and the compounds are expected to have attractive emission and absorption properties. The decrease in the energy of LUMO and HOMO orbitals is also responsible to cause the planarity in the molecule [47].
Conclusion
The reaction between thiophene-2,5-carbonyl dichloride and pyrazole derivatives in the presence of trimethylamine is an easy access to get further derivatives as described in this study. The compounds can act as tridentate (NSN) chelating ligands. In light of literature available the complexes of these ligands can show better photophysical properties.
References
Zhang TY, O’toole J, Proctor C (1999) Sulfur Rep 22:1–47
Isloor AM, Kalluraya B, Pai KS (2010) Eur J Med Chem 45:825
Mishra R, Jha K, Kumar S, Tomer I (2011) Der Pharma Chem 3:38
Radwan MA, Shehab MA, El-Shenawy SM (2009) Monat Chem 140:445
Sondhi SM, Jain S, Dinodia M, Kumar A (2008) Med Chem 4:146
Molvi KI, Vasu KK, Yerande SG, Sudarsanam V, Haque N (2007) Eur J Med Chem 42:1049
Scott AB, Elizabeth AM, Wei W, Michael W, Monica Justin A S, Michele S, Arminda B, Thomas K, David R, Dennis A (1997) Bioorg Med Chem 5:779
Wardakhan W, Abdel-Salam O, Elmegeed G (2008) Acta Pharm 58:1–14
Connor DT, Cetenko WA, Mullican MD, Sorenson RJ, Unangst PC, Weikert RJ, Adolphson RL, Kennedy JA, Thueson DO (1992) J Med Chem 35:958
Flynn BL, Verdier-Pinard P, Hamel E (2001) Org Lett 3:651
Charles DJ, Mary GJ, Andrew JP, Mary KP, Larry JB, Allen RT, Julie FF, James AC (1984) J Med Chem 27:1057
John DP, George DH, Pierre JM, Brian MM, Stuart RM, Mark AM, Harvey S, Robert LS, John MS, James PS, Michael FS (1991) J Med Chem 34:1805
Li X-C, Sirringhaus H, Garnier F, Holmes AB, Moratti SC, Feeder N, Clegg W, Teat SJ, Friend RH (1998) J Am Chem Soc 120:2206
Kim KH, Chi Z, Cho MJ, Jin J-I, Cho MY, Kim SJ, Joo J-S, Choi DH (2007) Chem Mater 19:4925
Merlo JA, Newman CR, Gerlach CP, Kelley TW, Muyres DV, Fritz SE, Toney MF, Frisbie CD (2005) J Am Chem Soc 127:3997
Yu C-Y, Chen C-P, Chan S-H, Hwang G-W, Ting C (2009) Chem Mater 21:3262
Köse ME, Mitchell WJ, Kopidakis N, Chang CH, Shaheen SE, Kim K, Rumbles G (2007) J Am Chem Soc 129:14257
Tsai J-H, Lee W-Y, Chen W-C, Yu C-Y, Hwang G-W, Ting C (2010) Chem Mater 22:3290
Chan HSO, Ng SC (1998) Prog Polym Sci 23:1167
Cheylan S, Fraleoni-Morgera A, Puigdollers J, Voz C, Setti L, Alcubilla R, Badenes G, Costa-Bizzarri P, Lanzi M (2006) Thin Solid Films 497:16
Huo L, Hou J, Zhang S, Chen HY, Yang Y (2010) Angew Chem Int Ed 49:1500
Chang YT, Hsu SL, Su MH, Wei KH (2009) Adv Mater 21:2093
Choi H, Paek S, Song J, Kim C, Cho N, Ko J (2011) Chem Commun 47:5509
Roland F, Egon R, Amaresh M, Elena M-O, Hannah Z, Christian K, Karl L, Moritz R, Mathias W, Olga T, André W, Christian U, Martin P, Peter B (2011) Adv Funct Mater 21:897
Bruno P, Hugo MS, Luz F, Berta C, Abel T, Emilia B, José LC, Teresa A, Carlos L (2007) Inorg Chem Commun 10:925
Mingqian H, Jianfeng L, Michael LS, Feixia Z, Robert RH, Hon HF, Vladimir AP, Detlef-M S, George GM (2009) J Am Chem Soc 131:11930
Renuga D, Udhayakumari D, Suganya S, Velmathi S (2012) Tetrahedron Lett 53:5068
Silvia VR, Nathaniel SF (2010) Org Lett 12:2598
Abdul RB, Asif IB, Fareeda A, Amir A (2009) Helvetica Chim Acta 92:1644–1656
Kotani S, Shiina K, Sonogashira K (1992) J Organomet Chem 429:403
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox (2010) Gaussian 09 (revision A.02), Gaussian Inc, Wallingford.
Axel DB (1993) J. Chem. Phys. 98:5648
Chengteh L, Weitao Y, Robert GP (1988) Phys. Rev. B 37:785
Stevens PJ, Devlin FJ, Chablowski CF, Frisch MJ (1994) J Phys Chem 98:11623
McLean D, Chandler DGS (1980) J Chem Phys 72:5639
Krishnan R, Blinkley JS, Seeger R, Pople JA (1980) J Chem Phys 72:650
Mohlala MS, Guzei IA, Darkwa J, Mapolie SF (2005) J Mol Catal A 241:93
Guzei IA, Spencer LC, Tshivashe MG, Darkwa J (2009) Acta Crystallogr E 65:2743
Kottke T, Stalke D (1993) J Appl Crystallogr 26:615
Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Crystallogr 32:115
Sheldrick GM (2008) Acta Cryst 64A:112
Farrugia LJ (1999) J Appl Crystallogr 32:837
Dahiya R, Pathak D (2007) Eur J Med Chem 42:772
Yüksektepe Ç, Çalişkan N, Genç M, Servi S (2010) Crystallogr Rep 55:1188
Debasish G, Ganesan M (2014) RSC Adv 4:45603
Ataf AA, Adnan S, Zarif G, Sher AK, Amin B, Muhammad NT, Zafar IZ, Ezzat K (2015) J Chem 2015:1–5. http://dx.doi.org/10.1155/2015/913435
Barbarella G, Favaretto L, Sotgiu G, Zambianchi M, Antolini L, Pudova O, Bongini A (1998) J Org Chem 63:5497
Acknowledgments
S.A.K. is grateful to Higher Education Commission (HEC) Pakistan for fellowship under “5000 indigenous PhD scholarships” at the University of Malakand and a 6 months fellowship under the “International Research Support Initiative Program” IRSIP as a graduate research trainee, Anorganische Chemie II, Universität Bayreuth, Bayreuth, Germany. We are also thankful to Prof. Dr. Rhett Kempe, Anorganische Chemie II, Universität Bayreuth, for his generous support in validating the crystal data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khan, E., Khan, S.A., Shahzad, A. et al. Synthesis Characterization and DFT Calculations of 2,5-Substituted Thiophene Derivatives. J Chem Crystallogr 45, 238–243 (2015). https://doi.org/10.1007/s10870-015-0588-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-015-0588-9