
Abstract
The binuclear zinc complex bis(2-bromobenzoato-O)-bis(μ 2 -2-bromobenzoato-O,O′)-bis(phenazone-O)-dizinc(II) (I) and the mononuclear dihydrate bis(2-bromobenzoato-O)-bis(thiourea-S)-zinc(II) (II) have been synthesized and characterized by means of elemental analysis and spectroscopic methods (IR, 1H and 13C NMR, EDS). The solid state structures of both compounds were determined by single-crystal X-ray diffractometry. Compound [Zn(2-BrC6H4COO)2(phen)]2 (phen—phenazone) (I) crystallized as a dimeric compound with a triclinic lattice (space group P − 1), where both zinc atoms, interconnected by two carboxylate groups, possess a distorted tetrahedral coordination environment. The crystallographic data of complex I: a = 9.9410(3) Å, b = 10.7309(3) Å, c = 12.9237(4) Å, α = 93.6004(17)°, β = 92.5898(11)°, γ = 116.2192(16)°, V = 1230.26(6) Å3, Z = 1. Complex [Zn(2-BrC6H4COO)2(tu)2]·2H2O (tu—thiourea) (II) crystallized with an orthorhombic lattice (space group Aba2) as a monomeric compound, where the coordination environment of the central zinc atom is a distorted tetrahedron. The crystallographic data of complex II are: a = 9.8595(3) Å, b = 19.7052(5) Å, c = 12.5908(3) Å, V = 2446.18(11) Å3, Z = 4. The modes of the carboxylate binding were assigned from the IR spectra using the magnitude of the separation between the carboxylate stretches (Δ), which correlated well with the crystal structures. The computed theoretical IR spectrum agreed well with the experimental data.
Graphical Abstract
Two new compounds bis(2-bromobenzoato-O)-bis(μ 2 -2-bromobenzoato-O,O′)-bis(phenazone-O)-dizinc(II) and bis(2-bromobenzoato-O)-bis(thiourea-S)-zinc(II) have been prepared and characterized by means of spectral analyses and X-ray single crystal structure determination; furthermore the correlation of IR spectra and structures of the prepared compounds has been done.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Zinc(II) carboxylates with O-, S-, N-donor ligands are interesting from several points of view. They exhibit antimicrobial properties and can be used for prevention and therapy of many illnesses in the form of components of drugs and biopreparations [1]. On the other hand, zinc(II) complexes are interesting also due to their variable structural properties since zinc is a metal very flexible in adopting various coordination geometries in its complexes. The majority of known complexes have a zinc atom with a tetra-, penta- or hexa-coordination environment around the central atom leading to distorted tetrahedral, square-pyramidal or octahedral geometries [2]. The carboxylate group has a versatile coordination behavior; it can exhibit ionic, monodentate or bidentate (bridging, chelating) binding mode to some metal ions [3].
In our previous works, we have studied physicochemical properties, crystal structures and biological activity of several aliphatic and aromatic zinc(II) carboxylates with bioactive ligands [1, 4–8]. The crystal structure of three aliphatic and one aromatic monomeric carboxylatozinc(II) complexes with thiourea is known from literature [9–12]. The last compound, namely benzoatozinc(II) complex with thiourea, contains two crystallographically different molecules, where both zinc atoms have tetrahedral arrangement formed by two oxygen atoms from two benzoate anions and by two sulfur atoms from two thiourea ligands [12]. Up to now, among the zinc(II) halogenobenzoates only the structure of 2-chlorobenzoatozinc(II) complex with phenazone has been determined. It has been observed that zinc(II) is coordinated in a distorted tetrahedral arrangement by two oxygen atoms from two phenazone ligands and two oxygens from two carboxylate anions [13]. Furthermore, the structural properties of some zinc(II) and copper(II) 2-bromobenzoates have been studied with several ligands, however with the notable exception of phenazone and thiourea [5, 7, 14–16].
The latter omission led us to initiate investigations of 2-bromobenzoatozinc(II) complexes with both the phenazone and the thiourea ligands.
Experimental
Materials
The following A. R. grade chemicals were used for the preparation of the studied compounds: ZnCl2 (Fluka), Na2CO3 (Mikrochem a.s. Pezinok), 2-bromobenzoic acid 97% (Aldrich), phenazone and thiourea (Merck) (Scheme 1).

a Phenazone (phen) and b thiourea (tu)
Syntheses
Compound [Zn(2-BrC6H4COO)2(phen)]2 (I)
25 cm3 of aqueous solution of ZnCl2 (1.36 g, 10 mmol) was added to 25 cm3 of aqueous solution of Na2CO3 (1.06 g, 10 mmol). The freshly prepared precipitate of ZnCO3 was purified from sodium chloride by decantation and added to the methanol solution of 2.58 g 97% 2-bromobenzoic acid. After stirring this mixture for 1 h at 50 °C, excess ZnCO3 was filtered off using a S4 frit. The filtrate was then mixed with 75 cm3 of aqueous solution of phenazone (2.26 g, 12 mmol) under continual stirring. The reaction solution was stirred further for 1 h at 50 °C and after reducing its volume to one half heating at 80 °C in a water bath it was left for crystallization at room temperature. After several days, bright square crystals of I were formed. The yield of the reaction was 72%. Anal. Calcd for C50H40O10N4Br4Zn2 (%): C, 45.90; H, 3.08; N, 4.29; Zn 10.00. Found: C, 46.28; H, 3.06; N, 4.35; Zn, 10.38. IR (KBr, cm−1): 3101m, 3068w [νar(C–H)]; 2995w, 2924w [\( \nu \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} \)]; 1647s [ν(C=O)]; 1601s, 1572s [νas(COO−)]; 1558m, 1458m [ν(C=C)]; 1498m [\( \delta_{\text{as}} \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} \)]; 1416s, 1377s [νs(COO−)]; 1315m [\( \delta_{\text{s}} \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} \)]; 1257w [ν(C–N)]; 1151w [ν(C–C)]; 646s [ν(C–Br)]; 750s [δar(C–H)]; 690m [δ(COO−)]. 1H NMR—(DMSO-d6, ppm): δ 2.23 (s, 6H, CH3), 3.05 (s, 6H, N–CH3), 5.30 (s, 2H, H-8′), 7.28 (m, 4H, H-4), 7.31 (m, 6H, H-2′,4′,6′), 7.38 (ddd, J = 7.6, 7.2, 1.0 Hz, 4H, H-5), 7.48 (m, 4H, H-3′,5′), 7.61 (dd, J = 7.2, 1.0 Hz, 4H, H-3), 7.65 (dd, J = 7.6, 1.6 Hz, 4H, H-6). 13C NMR—(DMSO-d6, ppm): δ 12.5 (N–CH3), 35.2 (CH3) 96.5 (C-8′), 119.5 (C-2), 123.5 (C2′,6′), 126.0 (C-4′), 127.1 (C-5), 128.9 (C-3′,5′), 130.2 (C-6), 130.4 (C-4), 133.0 (C-3), 135.3 (C-1′), 138.5 (C-1), 157.8 (C-7′), 165.42 (C=O), 171.4 (COO−).
Compound [Zn(2-BrC6H4COO)2(tu)2]·2H2O (II)
ZnCO3 was synthesized by the reaction of 25 cm3 of aqueous solution of 1.35 g (10 mmol) ZnCl2 and 25 cm3 of aqueous solution of 1.06 g (10 mmol) Na2CO3. The water suspension of the purified precipitate of ZnCO3 was added to the methanol solution of 2.58 g 97% 2-bromobenzoic acid. This mixture was stirred at 50 °C for 1 h and excess ZnCO3 was afterwards filtered off using an S4 frit. An aqueous solution of 0.912 g (12 mmol) thiourea was added to the filtrate. The reaction mixture was stirred at 50 °C for 1 h, its volume then reduced in a water bath at 70 °C and then left to crystallize at room temperature. After several days, the colorless crystalline compound of II was formed. The yield of the reaction was 84%. Anal. Calcd for C16H20O6N4S2Br2Zn (%): C, 29.38; H, 3.08; N, 8.57; S, 9.81 Zn, 10.00. Found: C, 29.84; H, 3.15; N, 8.94; S, 10.10; Zn, 10.79. IR (KBr, cm−1): 3614m [\( \nu \left( {\text{O}}{-}{\text{H}} \right)_{{{\text{H}}{}_{ 2}{\text{O}}}} \)], 3377s, 3315s [ν(N–H)]; 3142s [νar(C–H)]; 1649s [δ(N–H)]; 1630s [\( \delta \left( {\text{O}}{-}{\text{H}} \right)_{{{\text{H}}{}_{ 2}{\text{O}}}} \)]; 1587s [νas(COO−)]; 1510m, 1471m [ν(C=C)]; 1471m [ν(N–C–N)]; 1356m [νs(COO−)]; 762s [δar(C–H)]; 721m [ν(C=S)]; 698s [δ(COO−)]; 648s [ν(C–Br)]. 1H NMR—(DMSO-d6, ppm): δ 6.80–7.65 (bs, NH2, H2O), 7.28 (ddd, J = 7.8,7.3, 2.0 Hz, H-4), 7.37 (ddd, J = 7.7, 7.3, 1.1 Hz, H-5), 7.60 (dd, J = 7.8, 1.1 Hz, H-3), 7.63 (dd, J = 7.7, 2.0 Hz, H-6). 13C NMR—(DMSO-d6, ppm): δ 119.4 (C-2), 127.1 (C-5), 130.0 (C-6), 130.3 (C-4), 132.9 (C-3), 138.8 (C-1), 171.3 (COO−), 182.9 (C=S) (Scheme 2).

Numbering schemes of a 2-bromobenzoate and b phenazone used in interpretation of 1H and 13C NMR spectra
Instrumentation
The carbon, hydrogen and nitrogen contents in the synthesized compounds were determined by the CHN analyzer PERKIN ELMER 2400. The zinc contents have been determined using Complexone III as an agent and Eriochrome black T as an indicator.
Scanning electron microscope (SEM) analyses were done using Philips XL30 CP scanning electron microscope equipped with an energy dispersive spectroscopic (EDS) analyzer of X-ray radiation. The samples were placed on an adhesive C-slice coated with a 10 nm thick layer of Au/Pd alloy. The EDS analysis was performed using the EDAY Genesis Imaging/Mapping Program, version 3.605.
Infrared spectra were recorded on an AVATAR 330 FT-IR Thermo Nicolet spectrometer using KBr pellets (2 mg/200 mg KBr), in the range 4000–400 cm−1.
NMR spectra were obtained using a Varian Mercury Plus NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C at room temperature in hexadeuteriodimethyl sulfoxide. Tetramethylsilane was used as an internal standard for both nuclei (δTMS = 0.00 ppm). A series of standard 1D a 2D NMR experiments (1H, 13C, DEPT, H,H-COSY, gHSQC, gHMBC, NOESY) has been used to assign 1H and 13C signals. The heteronuclear HMBC experiment was optimized to 145 Hz (one-bond) and 5 Hz (long-range) J(H,C) couplings.
Density functional studies (DFT) have been carried out at the fermi cluster by using Gaussian 03, Revision E.01 [17]. All calculations used the Becke exchange [18] and the Perdew/Wang 91 correlational [19] functionals. For a proper elucidation of the theoretical infrared spectrum, the solid-state structure has been optimized using the 6-311 + G(3d,p) basis set [20] employed for all atoms and an analytical Hessian determined before the first step of the optimization procedure. Numerical integration was done on a pruned grid having 99 radial shells each of 590 angular points. The IR spectrum was computed after the optimization step yielded a proper stationary point where no imaginary frequencies were present. No scaling was employed for the bands computed.
Structure Determination and Refinement
Diffraction data were collected on a Nonius KappaCCD diffractometer (MoKα radiation, λ = 0.71073 Å) at 150(1) K and processed by the HKL program package [21]. The phase problem was solved by direct method (SIR97) [22], and the structure refined by full-matrix least-squares on F 2 (SHELX97) [23]. Hydrogen atoms were refined isotropically, all other atoms anisotropically. Hydrogen atoms residing on aromatic carbon atoms were included in ideal positions with C–H bonds fixed to 0.95 Å and Uiso(H) assigned to 1.2Ueq of the adjacent carbon atom; terminal methyl groups hydrogen atoms were included in ideal positions with C–H bonds fixed to 0.98 Å and Uiso(H) assigned to 1.5Ueq of the pivot atoms. The absorption correction was semi-empirical on symmetry equivalents. Important crystal data and refinement details for I and II are listed in Table 1.
Results and Discussion
Both complexes are stable on light and air at ambient temperature. Complex I is soluble in DMSO, DMF, H2O and CH3OH, slightly soluble in C2H5OH and insoluble in CHCl3, CCl4, (C2H5)2O and (CH3)2CO. Complex II is very well soluble in CH3OH, (CH3)2CO, DMF and DMSO, slightly soluble in C2H5OH and insoluble in CHCl3, CCl4 and H2O.
Spectroscopic Characterization
Compound [Zn(2-BrC6H4COO)2(phen)]2 (I)
The SEM micrographs of I showed a crystalline material consisting of plates 50–100 μm large and smaller particles exhibiting a broad particle size distribution. The chemical composition determined by EDS analysis was uniform. In all analyzed particles, every element expected in the proposed formula has been found (Supplementary material). The Zn:Br ratio determined by EDS analysis (1:2.1) corresponds well with the proposed formula within the accuracy limits of the EDS method.
The presence of the 2-bromobenzoato groups was apparent in the IR spectrum of the studied compound. The asymmetric and symmetric stretching vibrations of carboxylato groups were observed at νas(COO−) = 1601, 1572 cm−1 and νs(COO−) = 1416, 1377 cm−1, respectively. We suppose that bands at 1601 and 1377 cm−1 are due to the stretching vibrations of the two monodentate 2-bromobenzoate groups and the bands at 1572 and 1416 cm−1 due to stretches of two bridging 2-bromobenzoate anions. This is in accordance with literature data [4]. The presence of phenazone was confirmed by occurrence of the stretching and deformation vibrations of the CH3- group of the phenazone ligand (\( \nu \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} = 2 9 9 5 \) and 2924 cm−1, \( \delta_{\text{as}} \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} = 1 4 9 8\;{\text{cm}}^{ - 1} , \, \delta_{\text{s}} \left( {\text{C}}{-}{\text{H}} \right)_{{-{\text{CH}}_{ 3} }} = 1 3 1 5\; {\text{cm}}^{ - 1} \)). The strong absorption band of the carbonyl ν(C=O) vibration appeared at 1647 cm−1 and exhibited a shift to lower wavenumbers as compared with free phenazone (ν(C=O) = 1666 cm−1). This phenomenon can be explained by the coordination of the carbonyl oxygen to the central zinc atom, leading to a decrease of the double bond character of the carbonyl group and shifting the stretching vibration ν(C=O) to lower values. This assumption is in a good agreement with the results of the X-ray diffraction analysis. The IR data of I are listed in “Experimental” section.
Compound [Zn(2-BrC6H4COO)2(tu)2]·2H2O (II)
The chemical homogeneity of II with a roughly identical chemical composition of different crystals was showed by the EDS analysis. This analysis confirmed the presence of all elements corresponding to the suggested formula of the sample—C, N, O, Zn, Br and S (Electronic Supplementary Material). Taking into account the semiquantitative character of the EDS analysis, the results are in agreement with the proposed formula of the complex. The Zn:Br:S ratio, theoretically 1:2:2, was determined by EDS analysis as 1.0:2.0:1.9.
The solid state FT-IR spectrum confirmed the presence of functional groups of organic ligands. The presence of water manifested itself by the appearance of a broad band of stretching vibrations at 3614 cm−1 followed by vibration at 1630 cm−1 assignable to water deformation vibrations. The stretching vibrations of the thiourea N–H groups were found at 3377 and 3315 cm−1. The bands of asymmetric and symmetric stretching vibrations of COO− group occurred at 1587 and 1356 cm−1, respectively. IR spectra can also provide information regarding whether nitrogen or sulfur of thiourea is the atom involved in coordination. For the case when the sulfur atom of thiourea is the donor atom, the stretching vibration of C=S is shifted to a lower value compared to free thiourea. In II ν(C=S) appears at 721 cm−1. Compared with free thiourea (736 cm−1), the ν(C=S) shift to lower wavelengths is apparent, thus, the ligand coordination occurring via the sulfur atom was assumed. This conclusion was later confirmed by single crystal X-ray structure determination.
The comparison of experimental and theoretical values of the IR absorption bands appear in Table 2. These data are in good mutual agreement with the notable exception of the N–H stretching vibration which can be explained by the involvement of the N–H group in a solid-state hydrogen bridge. The N–H bond is thus prolongated, leading to the lowering in wavenumbers for this stretching vibration. This solid-state phenomenon could not be reflected by computational studies at all, since those could be carried out on one single molecule only.
Description of the Structure
Compound [Zn(2-BrC6H4COO)2(phen)]2 (I)
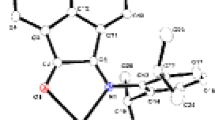
Complex I crystallized as a dimeric molecule with a triclinic lattice (space group P − 1), with no crystal water molecules present in its solid state structure (Fig. 1). The asymmetric part of the unit cell contains only one half of the dimeric molecule; the symmetrically dependent part is generated applying the crystallographic inversion centre, centered exactly on the interconnection between the two zinc atoms. Both zinc atoms have exactly the same coordination environment. The zinc atoms have a distorted tetrahedral coordination environment, consisting of four non-equivalent oxygen atoms bonded to one central atom. Three of these atoms are anions from three disparate carboxylate groups while the fourth atom is the ketone oxygen of the phenazone ligand. The distortion of the coordination environment around the central atoms is caused by the different steric demands of the individual ligands and possibly also by the distribution of the electrostatic potential over the whole molecule. Selected bond angles and distances are given in Table 3.

Solid state structure of I. Displacement ellipsoids are drawn at 30% probability level. Symmetrically dependant atoms are labeled with a terminal ‘i’. Hydrogen atoms are omitted for clarity. (i) –x + 1, –y + 1, –z + 1
This compound stabilizes its solid state arrangement via dimerization with two 2-bromobenzoate carboxylate groups serving as bridging groups between two metallic cations while no significant intermolecular hydrogen bridges are formed in solid state. Moreover, neither anion–π nor π–π interactions have been found to play an important role in the solid state packing.
Among carboxylatozinc(II) complexes, a similar coordination was found in benzoato (Parsons et al., Private communication, 2004) and 2-naphthyloxyacetato [24] complexes. The coordinate bond distances range from 1.939(3) to 1.973(2) Å (Table 3) which are slightly longer than those observed in [Zn(benzoato)2(6-methylpyridinio-2-olato)2]2 (from 1.912(2) to 1.972(3) Å [Parsons et al., Private communication, 2004]) and shorter than in [Zn(2-naphthylacetato)2(H2O)2]2 (from 1.949(3) to 2.014(4) Å [24]). Within the binuclear molecule the Zn···Zni distance 3.694(5) Å is shorter than those in the above-mentioned complexes, which exhibit Zn···Zni separations of more than 3.8 Å. This distance is longer than 3.247(3) Å in zinc(II) complex with three carboxylato bridges, [Zn(crotonato)2]2 [25] and in those with four carboxylato bridges: 2.8660(4) Å [Zn(benzoate)2(caffeine)2]2·2caffeine [26], 2.9697(4) Å in [Zn(2-bromobenzoato)2(N-methylnicotinamide)]2 [7] and 3.0533(4) Å in [Zn(benzoato)2(papaverine)2]2 [27].
Compound [Zn(2-BrC6H4COO)2(tu)2]·2H2O (II)
Complex II crystallized with orthorhombic lattice (space group Aba2) as a monomeric compound incorporating two crystal water molecules in solid state. The coordination environment of the central zinc atom is a distorted tetrahedron built from two oxygen atoms of two η1-coordinated 2-bromobenzoato groups and further from two sulfur atoms of two thiourea ligands (Fig. 2). The solid-state molecule possesses an exact twofold symmetry passing through the central zinc atom; the symmetrically dependant atoms of the complex together with the second crystal water molecule are obtained after this crystallographic twofold axis is applied. The distortion of the tetrahedron on the central atom is the consequence of unequal ionic radii of the coordinating sulfur and oxygen ions of both coordinated ligands. Selected bond angles and distances are included in Table 4. The primary stabilizing factor in the solid state arrangement is the presence of two crystal water molecules, both serving as linking agents between neighboring molecules through hydrogen bridges (Fig. 3, Table 5).

Solid state structure of II. Displacement ellipsoids are drawn at 30% probability level. Symmetrically dependant ligand atoms are labeled with a terminal ‘i’; the symmetrically dependant crystal water molecule is not included. (i) 1 − x, −y, z

Hydrogen bridges in the solid state structure of II. Hydrogen atoms of aromatic rings are omitted for clarity
The average Zn–O, Zn–S bond distances (2.0112(18) and 2.3256(8) Å) do not deviate markedly from those observed for the similar monomeric carboxylatozinc(II) complexes with thiourea: [Zn(CH3COO)2(tu)2] [1.964(9) and 2.29(3) Å [9]], [Zn(CCl3COO)2(tu)2] [2.006(6) and 2.304(13) Å [10]], [Zn(CH3CH2COO)2(tu)2] [1.971(17) and 2.333(10) Å [11]] and [Zn(C6H5COO)2(tu)2] [1.964(2) and 2.3673(14) Å; [12]]. Two additional Zn···O distances to the non-coordinated carboxylate O atoms are 2.829(2) Å (×2) and the highest value of tetrahedral angles X–Zn–Y is 119.62(11)°. The corresponding values of similar complexes containing ZnO2S2: 2.891(9) and 2.996(5) Å, 119.3(3)°; [9], 2.916(5) and 3.282(7) Å, 123.23(9)°; [10], 2.890(3) and 3.048(3) Å, 125.40(2)°; [11] and 2.957(3) (×2) and 2.955(3) (×2), 124.2(2) and 126.6(2)°; [12] are comparable with the obtained data. Černák et al. found a correlation between a type of donor atom (S, O, N) and the distance to the non-coordinated carboxylate O atom, as well as the degree of the tetrahedral deformation [12]. Our results are in good accordance with their observations.
Correlation of Spectral and Structural Properties
To correlate the infrared spectra with the structure of metal carboxylates, the difference between the asymmetric and symmetric carboxylate stretches (Δ = νas(COO−) − νs(COO−)) can be used. The following order has been proposed for divalent metal carboxylates [28, 29]:
The Δ (ionic) determined from IR spectra of sodium 2-bromobenzoate is 168 cm−1. For I the respective experimental values, Δexp, are 224 and 156 cm−1. The first value is an indication of the monodentate coordination of the carboxylate group as Δexp (224 cm−1) ≫ Δsodium 2-bromobenzoate (168 cm−1), while the other one of the bridging carboxylate coordination Δexp (156 cm−1) < Δsodium 2-bromobenzoate (168 cm−1). This result is in accordance with the crystal structure of I.
The relation between the values of Δ and the structure of carboxylate anion was proposed by Nara et al. [30] as: Δ = 1818.1δr + 16.47(OCO − 120) + 66.8, where δr is the difference between the two CO bond lengths (Å) and OCO is the OCO angle (°). This relation was applied for the case the compounds described in the present article, and after substitution of structural data (δr = 0.05 Å, OCO = 125.1(3)° (O9 C7 O8), and δr = 0.006 Å, OCO = 125.8(3)° (O18 C17 O19)), the calculated separations Δcalc = 241 cm−1 (O9 C7 O8) and 173 cm−1 (O18 C17 O19) have been obtained. For both cases, the Δexp values are inferior to the calculated ones. The comparison of the first value with the value Δ (ionic) = 168 cm−1 indicates a monodentate coordination, which is correct. On the other hand, the calculated value Δcalc = 173 cm−1 is slightly higher than Δ ionic, therefore this can not be assigned as a bridging coordination of carboxylate group. In the case of syn–syn bridges in zinc(II) benzoate with nicotinamide the Nara′s equation lost also its validity [4].
For II the separation of the stretches Δexp = 193 cm−1 indicates a monodentate coordination of the carboxylate group since Δexp (193 cm−1) ≫ Δsodium 2-bromobenzoate (168 cm−1). This result is in accordance with the determined crystal structure of II. The calculated value Δcalc from the Nara′s equation for structural data δr = 0.032 Å and OCO = 123.7(2)° is 186 cm−1 which agreed well with the experimental value (193 cm−1) .
Conclusion
Two novel 2-bromobenzoatozinc(II) compounds have been synthesized. The compound with phenazone was found to be dimeric crystallized with a triclinic lattice, where both zinc(II) atoms possess a distorted tetrahedral coordination environment (ZnO4). The compound with thiourea crystallized with an orthorhombic lattice as a monomeric compound including two crystal water molecules. The central zinc(II) atom is surrounded by a distorted tetrahedral coordination environment (ZnO2S2). It was found that the spectral and structural properties of the prepared compounds exhibit a good correlation.
Supplementary Material
CCDC 710294 and CCDC 710293 contain the crystallographic data of I and II for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge, CB2 1EZ, UK; fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk.
References
Győryová K, Szunyogová E, Kovářová J, Hudecová D, Mudroňová D, Juhászová E (2003) J Therm Anal Calorim 72:587
Melník M, Győryová K, Skoršepa J, Holloway CE (1995) J Coord Chem 35:179
Zeleňák V, Vargová Z, Győryová K (2007) Spectrochim Acta A66:262
Zeleňák V, Císařová I, Sabo M, Llewellyn P, Győryová K (2004) J Coord Chem 57:87
Erdélyiová A, Győryová K, Gyepes R, Halás L, Kovářová J (2009) Polyhedron 28:131
Findoráková L, Győryová K, Melník M, Koman M, Nour El-Dien FA (2010) J Coord Chem 63:3348
Krajníková A, Gyepes R, Győryová K (2010) J Chem Crystallogr 40:650
Bujdošová Z, Győryová K, Melník M, Koman M, Kovářová J (2011) J Chem Crystallogr 41:443
Cavalca L, Gasparri GF, Acreetti GD, Domiano P (1967) Acta Cryst 22:90
Potočňák I, Dunaj-Jurčo M, Petříček V, Černák J (1994) Acta Cryst C50:1902
Smolander K, Ahlgrén M, Melník M, Skoršepa J, Győryová K (1994) Acta Cryst C50:1900
Černák J, Adzimová I, Gérard F, Hardy AM (1995) Acta Cryst C51:392
Maroszová J, Findoráková L, Győryová K, Koman M, Melník M (2007) Acta Cryst E63:m1406
Hökelek T, Dal H, Tercan B, Özbek FE, Necefoglu H (2009) Acta Cryst E65:m607
Hökelek T, Dal H, Tercan B, Özbek FE, Necefoglu H (2009) Acta Cryst E65:m481
Moncol J, Maroszová J, Koman M, Melník M, Valko M, Mazur M, Lis T (2008) J Coord Chem 61:3740
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant, Millam JM, Iyengar SS, Tomasi S, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P,Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas Ö, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG,Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision E.01. Gaussian Inc, Wallingford
Becke AD (1988) Phys Rev A38:3098
Perdew JP, Burke K, Wang Y (1996) Phys Rev B54:16533
McLean AD, Chandler GS (1980) J Chem Phys 72:5639
Otwinowski Z, Minor W (1997) HKL Denzo and Scalepack Program Package. Nonius BV, Delft
Altomare A, Burla MC, Camalli M, Cascarano G, Giacovazzo C, Guagliardi A, Polidori G (1994) J Appl Crystallogr 27:435
Sheldrick GM (1997) SHELXL-97, program for X-ray crystal structure refinement. University of Göttingen, Gottingen
Chen FJ, Xu H, Xu H, Huang KL (2009) Acta Cryst E65:m540
Clegg W, Little IR, Straughan BP (1986) J Chem Soc Dalton Trans 1283:1986
Findoráková L, Győryová K, Koman M, Moncol J, Melník M (2010) J Chem Crystallogr 40:145
Zeleňák V, Sabo M, Massa W, Černák J (2004) Acta Cryst C60:m85
Deacon GB, Phillip RJ (1980) Coord Chem Rev 33:227
Nakamoto K (1997) Infrared and Raman spectra of inorganic and coordination compounds. Wiley, New York
Nara M, Torii H, Tasumi MJ (1996) Phys Chem 100:19812
Acknowledgments
This work was supported by the Slovak Ministry of Education (VEGA project 1/0122/08), the Ministry of Education of the Czech Republic (Long term research plan MSM0021620857) and the State NMR Program (Grant No. 2003SP200280203). The financial support is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Krajníková, A., Gyepes, R., Győryová, K. et al. Preparation, Crystal Structure and Spectroscopic Properties of Dimeric [Zn(2-bromobenzoato)2(phenazone)]2 and Monomeric [Zn(2-bromobenzoato)2(thiourea)2]·2H2O. J Chem Crystallogr 41, 1036–1043 (2011). https://doi.org/10.1007/s10870-011-0041-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-011-0041-7