
Abstract
The title compound was prepared by the reaction of 4-methyl-pent-3-en-2-one and malononitrile, at room temperature, in the presence of indium-triflate and triethylamine. The structure of the molecule was established by spectral analysis and X-ray diffraction studies. The compound crystallizes in the monoclinic space group C2/c with unit cell parameters: a = 14.269 (2), b = 10.141 (2), c = 19.743 (3) Å, β = 95.523(4)°, Z = 8. The crystal structure was solved by direct methods and refined to R = 0.0526 for 2,690 observed reflections. The isoquinuclidine rings adopt boat conformation. The molecules in the unit cell are arranged in layers. The crystal structure is stabilized by N–H⋯N interactions.
Graphical Abstract
Crystal structure analysis of the novel 2-amino-5-aza-6-(dinitrilomethylene)-4,7,7-trimethylbicyclo[2.2.2]octane-1,3-dicarbonitrile.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bicyclic, bridged, nitrogen-heterocycle scaffolds are found in a variety of naturally occurring bioactive alkaloids and synthetic products [1–6]. The muscarinic acid receptor [4–9] activity, associated with these compounds, makes them potential candidates for the treatment of ever increasing pulmonary and Alzheimers diseases [10, 11]. This has generated much interest for their synthesis. Azabicyclo [2.2.2] octanes, commonly referred as isoquinuclidines, have been synthesized by the Diels–Alder reaction of cyclohexenones and imines in the presence of chiral catalysts [12, 13]. Some enantioselective synthetic modifications of these reactions, involving the use of BINOL-derived phosphoric acids, have also been reported [14]. To the best of our knowledge, a direct synthesis of these compounds from acyclic aldehydes or ketones has not been reported so far. This prompted us to develop an appropriate methodology for the synthesis of polysubstituted azabicyclo[2.2.2]octanes, from the readily accessible acyclic ketones and malononitrile. In the course of these investigations, we obtained the title compound 1, in high yield (67%), by the one-pot reaction of 4-methyl-pent-3-en-2-one and malononitrile, in the presence of indium triflate and triethylamine. The structure of the compound was elucidated by spectral methods and XRD studies.

Experimental
Materials
All the materials used in the current study were obtained from Aldrich Chemicals, USA via Indian office.
Physical Measurements
Melting points are uncorrected and were determined on Perfit melting point apparatus. IR spectra were recorded on Bruker 4800 IR spectrometer. 1H NMR (200 MHz) and 13C NMR (50.3 MHz) spectra were recorded in (CD3)2CO, using a Bruker AcDPX-200 spectrometer; some spectra were recorded on Varian Gemini 300 MHz instrument. TMS was used as standard for NMR analysis. CHN analysis was done on Fison Model EA 1108 elemental analyzer. Thin layer chromatography was performed on 0.5 mm thick plates, using silica gel G (BDH) adsorbent. Column chromatography was performed on silica gel (mesh size 60–120 BDH).
Synthesis
4-Methyl-pent-3-en-2-one (1 × 10−2 mol) and malononitrile (3 × 10−3 mol), in acetonitrile (10 mL), were stirred, at room temperature, in the presence of indium triflate (20 mol %) and triethylamine (1.5 mL). After stirring for 24 h light yellow crystals of the title compound separated out. The reaction mixture was filtered, washed with cold acetonitrile (5 mL) and dried. Compound 1 was obtained in 67% yield. For the analytical purposes the compound (500 mg) was purified by column chromatography, on silica gel, using chloroform–ethyl acetate (19:1 v/v) as solvent, followed by crystallization from methanol–chloroform. For XRD study single crystals were obtained by slow evaporation of the methanol solution, at room temperature. The compound melted at 215 °C (decomp.).The compound gave the following analytical and spectral data.
Anal: CHN (%): Found. C, 64.81; H, 5.10; N, 30.27 (calc. for C15H14N6: C, 64.7; H, 5.0; N, 30.2). IR (KBr): υ max 3331 (NH2), 3243 (NH), 2195 (d), 1650, 1618, 1468, 1469, 1442, 1102, 1060, 874 cm−1. 1H-NMR (CD3)2CO: δ H 1.42 (3H, s), 1.43 (3H, s), 1.67 (3H, s), 1.72 (1H, d, J = 15 Hz), 1.88 (1H, d, J = 15 Hz), 7.42 (1H, s br, exch. D2O), 10.23 (1H, s br, exch. D2O). 13C-NMR (CD3)2CO: δ C 22.8, 26.8, 27.1, 38.4, 42.9, 48.3, 56.3 (CH2), 57.7, 79.4, 112.04, 114.1, 115.5, 115.7, 154.5,163.5.
Crystal Structure Determination and Refinement
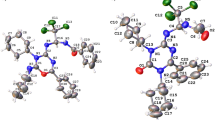
X-ray intensity data of 8,917 reflections (of which 3,461 unique) were collected at 100 K on Bruker CCD area-detector diffractometer equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å). The crystal used for data collection was of dimensions 0.3 × 0.2 × 0.1 mm. The cell dimensions were determined by least-square fit of angular settings of 2,715 reflections in the θ range 2.47–28.14°. The intensities were measured by ϕ and ω scan mode for θ ranges 2.47–28.29°. 2,690 reflections were treated as observed (I > 2σ(I)). Data were corrected for Lorentz and polarisation factors. The structure was solved by direct methods using SHELXS97 [15]. All non-hydrogen atoms of the molecule were located in the best E-map. Full-matrix least-squares refinement was carried out using SHELXL97 [15]. All the hydrogen atoms were located on a difference electron density map and their positional and isotropic thermal parameters were included in the refinement. The final refinement cycles converged to an R = 0.0526 and wR (F 2) = 0.1276 for the observed data. Residual electron densities ranged from −0.270 to 0.368 e Å−3. Atomic scattering factors were taken from International Tables for X-ray Crystallography (1992, Vol. C, Tables 4.2.6.8 and 6.1.1.4). The crystallographic data are summarized in Table 1. An ORTEP view of the title compound with atomic labeling is shown in Fig. 1 [16]. CCDC—750140 contains the supplementary crystallographic data for this paper.

ORTEP view of the molecule, showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii
Results and Discussion
Synthesis
Indium salts are established versatile Lewis acids for organic reactions and their utility in organic synthesis has been reviewed [17–19]. We became intrigued by the possibility that in the presence of indium triflate and triethylamine, ketones and malononitrile may undergo a cascade sequence of reactions involving both carbonyl and nitrile groups. The tendency of triethylamine to extract a proton from an activated carbon and effective ligand formation may facilitate the addition, under mild conditions, on the otherwise difficult nitrile group. Indeed our presumption worked well. Acetone and malononitrile, on stirring with indium triflate and triethylamine, at room temperature, formed compound 1 (C15H14N6), which separated out as light yellow crystals. After purification by column chromatography and crystallization the compound was obtained in 67% yield. The structure of the compound was elucidated by spectral methods: IR, 1H-NMR, 13C NMR, Distortionless Enhancement be Polarization Transfer (DEPT 135°), Heteronuclear Multiple Bond Coherence (HMBC) and Heteronuclear Multiple Quantum Coherence (HMQC). The IR spectrum of the compound contained prominent absorption bands at υ max cm−1 3331 (NH2), 3243 (NH), 2195 (d, CN), besides other expected absorption bands. The 1H-NMR spectrum accounted for three tertiary methyls at δ H 1.42 (3H, s), 1.43 (3H, s), 1.67 (3H, s) and a methylene group at δ H 1.72 (1H, d, J = 15 Hz), 1.88 (1H, d, J = 15 Hz). The presence of four nitrile groups was confirmed by the 13C NMR signals at δ c 112.04, 114.1, 115.5, 115.7. DEPT 135°, HMBC and HMQC experiments confirmed the presence of lone methylene group, δ c 56.3, whose protons were coupled to each other. The presence of three quartets in the HMBC spectrum, δ c 22.8, 26.8, 27.1, substantiated the presence of three tertiary methyls in the compound. The resonance signals at δ c 154.5, 163.5 were assigned to the double bonded carbons bearing nitrile and amino group, respectively. The analytical studies and spectral data of the compound revealed its structure as 2-amino-5-aza-6-(dinitrilomethylene)-4,7,7-trimethylbicyclo[2.2.2]octane-1,3-dicarbonitrile.
Mechanistically, the reaction seems to involve aldol-type condensation followed by Michael addition. Subsequent addition to nitrile group and annulation reaction (Scheme 1) may lead to compound 1.

Probable mechanism for formation of the title compound
The XRD studies showed that in compound 1 bond lengths and bond angles are in agreement with those reported for other structure determinations of isoquinuclidine systems [20, 21]. The mean bond length of nitrile groups is 1.147 (2) Å, which is comparable with the values reported for nitrile-substituted organic ligands [22]. The two triangular isoquinuclidine planes, viz. C2–C6–C7 and C3–N5–C8, define a dihedral angle of 5.60 (5)°, and are thus tilted slightly with respect to their parallel position in the ideal conformation. The geometry of exocyclic dinitrilo methane group shows that only one conjugated nitrile group C12 is linear while the second nitrile group C11 is marginally deviated from linearity. The C10–C12–N20 bond angle is 179.02 (15)° whereas C10–C11–C19 angle is 173.94 (15)°. The C1–C6–C10 bond angle is 126.24 (12)° much higher than 120°. This may be attributable to the repulsive interactions between the bridge head nitrile C9 and C11 nitrile groups which are in proximity to each other.
Ring A [C1–C4, N5, C6] has a boat conformation with C1 and C4 −0.688 (1) and −0.650 (1) Ǻ, respectively, from the C2, C3, N5, C6 plane. The asymmetry parameter ΔC s (C1 − C4) = 0.72 [23]. The conformation of ring B [C1–C4, C8, C7] is boat, with asymmetry parameter: ΔCs (C1 − C4) = 5.60. Atoms C1 and C4 are situated 0.782 (1) and 0.699 (1) Ǻ, respectively, above the plane defined by the other four ring atoms. The conformation of ring C [C1, C7, C8, C4, N5 C6] is also boat [ΔCs(C1 − C4) = 5.27]. Atoms C1 and C4 are situated 0.730 (1) and 0.717 (1) Ǻ, respectively, above the plane defined by the other four ring atoms.
Packing view of the molecules in the unit cell viewed down the a axis is shown in Fig. 2. Molecules in the unit cell are packed together to form well defined layers. The crystal packing involves N–H⋯N hydrogen bond interactions. Amino atom N5 at (x, y, z) acts as donor to carbonitrile nitrogen atom N20 at (−x, y, ½ − z). In addition amino atom N17 at (x, y, z) acts as donor to two carbonitrile nitrogen atoms N19 at (½ − x, −½ − y, 1 − z) and N21 at (1 − x, y, ½ − z). The geometry of N–H⋯N hydrogen bonds is given in Table 2. To the best of our knowledge this compound has not been reported so far. Further scope of this reaction is under investigation and will be communicated to press in due course of time.

The crystal packing projected on to the bc plane
Conclusion
For the first time, 2-amino-5-aza-6-(dicyanomethylene)-4,7,7-trimethylbicyclo[2.2.2]-octane-1,3-dicarbonitrile was prepared, in high yield, by the reaction of acetone and malononitrile, in the presence of indium-triflate and triethylamine, under ambient reaction conditions. The isoquinuclidine skeleton adopts boat conformation and the molecules in solid state are stabilized by N–H⋯N hydrogen bonding.
Supplementary Material
CCDC—750140 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
O’Hagan D (2000) Nat Prod Rep 17:435
Weiland H, Schopf C, Hermsen W (1925) Liebigs Ann Chem 444:40
Kanistan HU, Garner P (2007) J Am Chem Soc 129:15460
Cereda E, Ezhaya A, Gil QM, Bellora E, Dubini E, Micheletti R, Shiavone A, Brambilla A, Shiavi GB, Donetti A (1990) J Med Chem 33:2108
Godovickov NN, Dorofeeva NA, Kardanov NA, Trifonova SA, Shelkovnikov SA (1985) Khim Farm Zh 19:1060
Tropsha AE, Nizhnii SV, Iaguzhinskii LS (1985) Bioorg Khim 11:1402
Rzeszotarski WJ, McPherson DW, Ferkany JW, Kinnier WJ, Naronha-Blob L, Rzeszotarski AK (1998) J Med Chem 31:1463
Kiesewetter DO, Silverton JV, Eckelman WC (1995) J Med Chem 38:1711
Martel AM, Rabasseda X, Castaner J (1997) Drugs Future 22:135
Gross NJ (2006) Eur J Pharmacol 533:36
MacLeeod AM, Merchants KJ, Shewell GA, Saunders J, Herbert RH, Freedman SB, Harley EA (1990) J Med Chem 33:2690
Sunden H, Ibrahem H, Erickson L, Cordova A (2005) Angew Chem Int Ed 44:4877
Liu H, Cun L-F, Mi A-Q, Jiang Y-Z, Gong L-Z (2006) Org Lett 8:6023
Rueoing M, Azap C (2005) Angew Chem Int Ed 44:7832
Sheldrick GM (1997) SHELXS97 and SHELXL97. University of Gottingen, Gottingen
Farrugia LJ (1997) J Appl Crystallogr 30:565
Kumar S, Kaur P, Kumar V (2005) Curr Org Chem 9:1205
Fringuilli F, Piermatti O, Pizzo F, Voccaro L (2003) Curr Org Chem 7:1661
Zhang Y, Li P, Wang M (2009) J Org Chem 74:4364
Vencato I, Mascarenhas YP, Braz FR (1987) Acta Crystallogr C43:762
Moisan L, Doris E, Rousseau B, Thuery P (2004) Acta Crystallogr C60:792
Zhang XM, Lu JT (2007) Acta Crystallogr E63:3861
Duax WL, Norton DA (1975) Atlas of steroid structures, vol 1. Plenum, New York
Acknowledgments
The authors are thankful to Prof. P. K. Bharadwaj, Department of Chemistry, IIT, Kanpur, India, for the use of the data collection facility.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mahajan, N., Gupta, V.K., Kotwal, P. et al. An Efficient and Simple One-Pot Synthesis of Novel 2-Amino-5-aza-6-(dinitrilomethylene)-4,7,7-trimethylbicyclo[2.2.2]octane-1,3-dicarbo-nitrile and its Crystal Structure. J Chem Crystallogr 41, 552–556 (2011). https://doi.org/10.1007/s10870-010-9919-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-010-9919-z