
Abstract
A new hybrid phosphate, (C6H5NH3)[ZnCl(HPO3)], has been synthesized and its structure characterized from single-crystal X-ray diffraction. The title compound crystallizes in the orthorhombic space group Pbca (n. 61) with the unit-cell parameters: a = 9.8635(2) Å, b = 9.4516(10) Å, c = 22.2430(4) Å, Z = 8 and V = 2,073.62(6) Å3. The final R factors were R/ωR = 0.0361/0.0924. Its framework might be described as a layered structure with two (010)-parallel cationic and anionic layers. The IR spectrum of this phase shows characteristic bands of phosphite and anilinium groups.
Index abstract
The crystal structure is described as a layered one with two (010)-parallel layers types: Anionic, made from edges and/or corners sharing of [ZnO3Cl] and [HPO3], and cationic build upon isolated [(C6H5NH3)]+ cations.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The survey of the literature has shown that in recent years great interest has been focused on the organically templated compounds. In fact, these materials attracted considerably the attention of researchers due to especially their interesting optical properties [1]. Moreover, these hybrid materials are highly attractive for application to frequency doubling of the light produced by semiconductor lasers [2]. They are also interesting for their potential applications in different fields: biology, catalytic, electrical conductivity, magnetism, photochemistry and even as adsorbents and ions exchangers [3–7].
A large number of these hybrid phosphates have been reported in the literature: (NC5H12)2Zn3(HPO3)4 [8], [H2N(CH2)2NH2]0.5·ZnHPO3 [9], [CH3CH(NH3)CH2NH3][Zn2(HPO3)3]·H2O and [H3N(CH2)6NH3]·[Zn3(HPO3)4] [10], Zn3(HPO3)4·Ni(en)2(H2O)2 [11] (en for ethylenediamine), (C2H10N2)[Co3(HPO3)4] [12], (C2H10N2)[Mn3(HPO3)4] [13] and (C3H12N2)[Mn3(HPO3)4] [14].
We report in this work on a new layered Inorganic–Organic Hybrid Anilinium Zinc(II) Chloride Phosphite [(C6H5) NH3]+ [ZnCl(HPO3)]−, with an elucidated crystal structure and IR study.
Experimental
To prepare crystals of the title compound we proceeded as follow: 5.0588 g (54 mmol) of aniline and 1.3628 g (10 mmol) of ZnCl2 were added to 10 ml of aqueous H3PO3 (1 M) solution. The mixture was stirred for 8 h and then allowed to stand, at room temperature, for 2 weeks. The colourless hexagonal shaped crystals were filtered off and washed with an aqueous 80% ethanol solution.
Single Crystal Study
The X-ray diffraction data for a selected crystal were collected with a Nonius Kappa CCD area detector diffractometer using a monochromatic MoKα radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effect, and for adsorption effect [15].
The structure was solved by the Direct Methods procedure of SIR97 [16], and refined by the full matrix least square technique of SHELX-L 97 [17]. All the hydrogen atoms were located from difference Fourier maps.
Experimental details and the crystallographic data are reported in Table 1. Atomic coordinates of atoms in the structure are reported in Table 2, while Table 3 reports the most significant bond distances and angles. Supplementary data, like full bond lengths and angles and anisotropic displacement parameters, have been deposited at the Cambridge.
Crystallographic Data Centre and allocated the deposition number CCDC 289610 [The Cambridge Crystallographic Data Centre, deposit@ccdc.cam.ac.uk http://www.ccdc.cam.ac.uk/deposit. Telephone: (44) 01223 762910 Facsimile: (44) 01223 336033 Postal Address: CCDC, 12 Union Road, CAMBRIDGE CB2 1EZ, UK].
Infrared Spectrum
Infrared spectrum of the title compound was recorded, as suspension powder in KBr, on a Perkin-Elmer Spectrometer 1750, in the range 4,000–400 cm−1. Table 5 reports the band assignments for the IR spectrum.
Structure Description
The crystal structure of (C6H5NH3)[ZnCl(HPO3)] might be described by double layers scheme. As can be evidenced from Figs. 1 and 2, the framework of the title compound can be regarded as a layered structure with two (010)-parallel layers types: (1) the first layer (A) results from edges and/or corners sharing of [ZnO3Cl] and [HPO3] tetrahedra forming the unit [ZnCl(HPO3)]−, this is an anionic layer; (2) the second family of layers (C), is an organic layer made exclusively of [(C6H5NH3)]+ cations. Layers A and C are linked via H bonds interactions, reported as dashed lines on Fig. 1.

Crystal structure of (C6H5NH3)[ZnCl(HPO3)], projection down a-axis (H-bonds as dashed lines)

Projection down b-axis, a layered structure
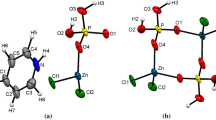
In the anionic layer, Zn2+ possesses a tetrahedral coordination: three oxygen atoms, from three different phosphites groups, and a chlorine atom. Figure 3 depicts the coordination around P and Zn, together with the conformation of C6H5NH3. Average distance d(Zn–O) of 1.9467 Å is similar to that observed in some other Zn-hybrid phosphates : (NC5H12)2Zn3(HPO3)4 [d(Zn–O) = 1.939(2) Å] [8], [H2N(CH2)2NH2]0.5·ZnHPO3 [d(Zn–O) = 1.934(1) Å] [9] and [CH3CH(NH3)CH2NH3][Zn2(HPO3)3].H2O [d(Zn–O) = 1.937 (3) Å] [10]. And then d(Zn–Cl) = 2.2296(8) Å, a value to compare with 2,2649(5) Å in (NH4) 3[ZnCl4]NO3 [18] or 2,2619(5) Å in K3[ZnCl4]NO3 [18].

Asymmetric unit of the title compound with completed P and Zn coordination. Displacement ellipsoids are drawn at the 50% probability level
The geometry of the hydrogenphosphite group (HPO3) is unexceptional (Fig. 2). Average distances dav(P–O) = 1.5153 (16) Å and dav(P–H) = 1.26 (3) Å are in good agreement with the values reported from other X-ray studies of known phosphite salts [9 and 19]. The O–P–O bond angles are in the range 107.4(9)–115.70(8)°, which are typical values for this ion. The H atom bonded to phosphorus is not involved in any hydrogen bonding. The average values of H–P–O, O–P–O and Zn–O–P angle are, respectively 106.6(12), 112.1(1) and 134.15(10)°. These values are in similar to those reported [8 and 9]. The [ZnO3Cl] and [HPO3] tetrahedrons share corners to delimit two kinds of cavities: hexagonal and rectangular along the direction [001] (see Fig. 4).

Projection of the structure of (C6H5NH3)[ZnCl(HPO3)] down c.: hexagonal and rectangular cavities
The intramolecular distances and angles of the anilinium cation: C(1)–N (1.462(3) Å), C–C (1.378(6) Å), C–H (1.05(6) Å), N–H (0.85(4) Å) and C(6)–C(1)–C(2) (122.5 (3)°) are in good agreement with those given in literature [20–22]. As in some other anilinium based compounds, the symmetry of the benzene ring is closer to C2v (mm) [23]. Both distances and angles are highly distorted in the ring. The two aromatic C–C bonds involving the C atom ipso to the substituent, C1–C2 and C1–C6, are somewhat shorter than the central C–C bonds of the ring, C2–C3 and C5–C6. The C1–N distance is close to the lower band of the range reported for anilinium salts with an average of 1.462(3) Å, close to the value reported in (C6H5NH3)[H2PO3], 1.465(7) Å [24].
The NH3+ group is staggered with respect to the phenyl ring. The aniline cations [(C6H5NH3)]+ are positioned between the layers to compensate the negative charges (Fig. 1) with the hydrogen bond N-H102–Cl with the value is 2.230(3) Å.
An intricate hydrogen bonding scheme contributes to build up the framework. The strongest Hydrogen bonds are the ones donated by nitrogen atom toward the phosphate group oxygens, displaying the shortest Donor–Acceptor distance of 2.772(3) Å (N1–H101…O3 x+1/2, −y−1/2; −z+1).
The eight strongest Hydrogen bonds are reported in Table 4. Representative of each kind of these H-bond interactions are reported on Fig. 1 as dashed lines.
Infra-Red Spectrum of [(C6H5)NH3]+ [ZnCl(HPO3)]−
Figure 5 depicts the IR spectrum of the title compound, while Table 5 summarises the attributions of the different bands in this spectrum. The later shows the presence of vibrational bands at 2,364, 2,388 and 2,416 cm−1 characteristic for stretching vibration of P–H and bands at 1,150, 1,000 and 483 cm−1 that are assigned to the vibration for a PO3 group.

IR spectrum of (C6H5NH3)[ZnCl(HPO3)]
The asymmetric and symmetric stretching vibrations of NH3 group are observed at 3,541 and 3,220 cm−1, and the bands observed at 1,617 and 1,636 cm−1 are due to the asymmetric deformation of the group NH3. The symmetric deformation of the NH3 group contributes a very strong absorption band at 1,573 cm−1. The stretching vibration of C–N is observed at 1,206–1,220 cm−1 and at 559 cm−1.
The CH stretching vibration is usually absorbing between 3 and 120–3,000 cm−1. In the present study, the band at 3,048 cm−1 is assigned to the CH stretching and the strong absorption band at 1,499 cm−1 and the weak component at 1,468 cm−1 are due to the ring stretching vibrations 13a(A1) and 13b(B2). The bonding vibrations of CH in and out of plane are observed, respectively at 1,029–619 cm−1. The bands at 749 cm−1 and at 1,147 cm−1 are assigned, respectively to the deformation vibration, in and out, of the plan for CH.
References
Batten SR, Robson R (1998) Chem Int Ed Engl 37:1460
Carter RL, Zompa L (1999) J Acta Cryst C55:6
Clearfield A (1998) Chem Mater 10:2801
Davis ME, Lobo RF (1992) Chem Mater 4:756
Domenicano A, Serantoni EF, Sanseverino LR (1977) Acta Cryst B33:1664
Fernandez S, Mesa JL, Pizarro JL, Lezama L, Arriortua MI, Olazcuaga R, Rojo T (2000) Chem Mater 12:2092
Fernandez S, Pizarro JL, Mesa JL, Lezama L, Arriortua MI, Rojo T (2001) Int J Inorg Mater 3:331
Fernandez S, Mesa JL, Pizarro JL, Lezama L, Arriortua MI, Olazcuaga R, Rojo T (2001) Inorg Chem 40:3476
Fu W, Shi Z, Zhang D, Li G, Dai Z, Chen X, Feng SJ (2003) Solid State Chem 174:11
Gao X, Davies JP, Weaver MJ (1990) J Phys Chem 94:6858
Gordon LE, Harrison WTA (2003) Acta Cryst E59:o195
Harrison WTA (2001) J Solid State Chem 160:4
Idrissi AK, Rafiq M, Gougeon P, Guerin R (1995) Acta Cryst C51:1359
Kondo T, Ogasawara N, Ito R, Ishida K, Tanase T, Murata T, Hidai M (1988) Acta Cryst C44:102
Lin ZE, Zhang J, Zheng ST, Yang GY (2004) Eur J Inorg Chem 953
Paixão JA, Matos Beja A, Ramos Silva M, Martin-Gil J (2000) J Acta Cryst C56:1132
Rodgers JA, Harrison WTA (2000) Chem Commun 2385
Sheldrick GM (1996) SADABS, Absorption Correction Program. Göttingen University, Germany
Sheldrick GM (1997) SHELXL-97. Program for the Refinement of Crystal Structures. Göttingen University, Germany
Shi Z, Feng S, Zhang L, Yang G, Hua J (2000) Chem Mater 12:2930
SIR97–Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Cryst 32: 115
Tordjman I, Masse R, Guitel JC (1988) Acta Cryst C44:2055
Zaccaro J, Bagieu-Beucher M, Espesso J, Ibanez AJ (1998) Cryst Growth 186:224
Zaworot MJ (1998) Chem Int Ed Engl 37:1211
Acknowledgments
BEB wishes to thank Dr M. Dusek [Institute of Physics, Praha, Czeck Republic] for his unquestioning collaboration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chaouche, S., Ouarsal, R., Lachkar, M. et al. Crystal Structure and IR Study of (C6H5NH3)[ZnCl(HPO3)]. J Chem Crystallogr 40, 486–490 (2010). https://doi.org/10.1007/s10870-009-9682-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-009-9682-1