
Abstract
The tridentate Schiff base carboxylate-containing ligand 2-pyridylmethylamino-3-butyric acid (Hpaba) reacts with copper(II) perchlorate to give the one-dimensional chain complex {[Cu(μ-paba)](ClO4)·H2O} n (1). The coordination geometry of each copper(II) ion is best described as a square-plane with two nitrogen atoms and one carboxylate oxygen atom of the ligand, and one carboxylate oxygen atom belong to another ligand. It crystallizes in the monoclinic system P21/c with a = 12.717(2), b = 7.9163(15), c = 14.981(2) Å, β = 111.14(1)°, V = 1406.8(4) Å3, Z = 4. Cyclic voltammogram of 1 undergoes the reversible one-electron oxidation to the Cu(III) and the reversible one-electron reduction to the Cu(I) state.
Graphical Abstract
Crystal structure of 1 reveals a syn-anti carboxylate-bridged one dimensional chain complex in which the coordination environment around each copper(II) ion exhibits a distorted square plane.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Self-assembled coordination polymers of transition metal(II) ions and Schiff base ligands have been of great interest due to their importance as models for biologically active sites [1–5], catalysts [6, 7], optical properties [8, 9], and magnetism [10–16]. It is well known that the structures and chemical properties of these compounds can be influenced by the metal ion, the structure of Schiff base ligands, the possible counterion and solvent effects. For example, the self-assembly of copper(II) perchlorate with bis(2-pyridylmethyl)amino-4-butyric acid (Hpmba) leads to the carboxylate-bridged one-dimensional chain complex {[Cu(μ-pmba)(H2O)](ClO4) · 3H2O} n , which exhibits a distorted square-pyramidal geometry with a N3O basal plane and a water molecule in the axial position [15]. However, copper(II) chloride reacts with 2-(imidazolemethyl)amino-3-propionic acid (Hiap) to give the chloride-bridged dinuclear square-pyramidal complex [Cu(Hiap)(μ-Cl)]2 [14]. In this case, the bridged chloride anion prevents a self-assembly reaction through the carboxylate group. On the other hand, the reaction of copper(II) chloride with Hpmba ligand leads to the mononuclear square-pyramidal complex [Cu(Hpmba)Cl2]·H2O] [17] in which the presence of chloride ions in the reaction mixture prevents any dimmer and chain formation. In order to better understand some aspects of different molecular topologies, we prepared the one-dimensional copper(II) complex {[Cu(μ-paba)](ClO4) · H2O} n (1) (Hpaba = 2-pyridylmethylamino-3-butyric acid). The Hpaba ligand does not saturate the coordination positions on the copper(II) ion. It was thought that a self-assembly reaction may take place through the deprotonated carboxylate group, leading to a syn–anti carboxylate-bridged one-dimensional chain.

Experimental
Materials and Physical Measurements
All chemicals used in syntheses were of reagent grade and were used without further purification. IR spectra were recorded with a Perkin-Elmer Paragon 1000 FT-IR spectrophotometer using KBr pellets. The solution electronic and diffuse reflectance spectra were obtained on a Jasco V-550 spectrophotometer. Elemental analyses were performed on a Perkin-Elmer CHN-2400 analyzer. Electrochemical measurements were accomplished with a three electrode potentiostat BAS-100BW system. A 3-mm Pt disk was used as the working electrode. The counter electrode was a coiled Pt wire and a Ag/AgCl electrode was used as a reference electrode. Cyclic voltametric data were obtained in DMSO solution using 0.1 M tetraethylammonium perchlorate (TEAP) as supporting electrolyte at 20.0 ± 0.1 °C. The solution was degassed with high purity N2 prior to carrying out the electrochemical measurements.
Synthesis of 2-Pyridylmethylamino-3-butyric acid (Hpaba)
To water solution (25 mL) of 2-picolyl chloride hydrochloride (1.64 g, 10 mmol) was added a water solution of 3-aminobutylic acid (1.03 g, 10 mmol) and sodium hydroxide (0.4 g, 10 mmol). After the mixture was heated to reflux for one day, the solution was extracted with chloroform. The removal of the solvent yielded the crude white product, which was purified by recrystallization from a 1:1 water–ethanol mixture (10 mL, v/v). Yield 1.52 g (78%). Calc. (found) for C10H14N2O2: C, 61.84 (61.76); H, 7.27 (7.35); N, 14.42 (14.36)%.
Synthesis of {[Cu(μ-paba)](ClO4) · H2O} n (1)
A methanol solution (20 mL) of Cu(ClO4)2 · 6H2O (185 mg, 0.5 mmol) and Hpaba (97 mg, 0.5 mmol) was heated to reflux for 1 h and then cooled to room temperature. The solution was filtered and left at room temperature until the blue crystals formed. The product was filtered and recrystallized from a hot water/acetonitrile (1:1, 10 mL) mixture. Yield: 135 mg (72 %). Calc. (found) for C10H15ClCuN2O7: C, 32.09 (32.15); H, 4.04 (4.12); N, 7.49 (7.41)%. IR (KBr, cm−1): 3448(m), 3237(m), 1615(s), 1593(s), 1575(w), 1532(s), 1489(m), 1448(s), 1406(m), 1292(m), 1101(s), 1034(m), 946(w), 774(m), 724(w), 660(w), 624(s), 424(m). UV–vis in acetonitrile [λmax, nm (ε, M−1 cm−1)]: 715 (120), in DMSO: 714 (118); in diffuse reflectance (λmax, nm): 712.
X-ray Crystallography
Intensity data for 1 were collected on an Enraf-Nonius CAD4 diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) in the ω − 2θ scan mode. Accurate cell parameters and an orientation matrix were determined by least-squares fit of 25 reflections. The intensity data were corrected for Lorentz and polarization effects. Absorption correction was applied by φ-scan [18]. The structure was solved by direct method [19] and the least-squares refinement of the structure was performed by the SHELXL97 package program [20]. All non-hydrogen atoms except for the perchlorate anion were smoothly refined, but four oxygen atoms, O(3), O(4), O(5), and O(6) of the perchlorate group vibrated so severely that their isotropic thermal parameters were abnormally big. All hydrogen atoms except for two water hydrogen atoms were placed in calculated positions, allowing them to ride on their parent C atoms, with Uiso (H) = 1.2 Ueq (C). When the structure was refined anisotropically, all of the four oxygen atoms were disordered over two positions as guessed. Therefore, they were divided into two groups A and B, and their positions including their occupancy factors were anisotropically refined using PART instruction. The final occupancy factors were 0.43 and 0.57 for the groups A and B, and the oxygen atoms O(6A), O(4B), O(6B) were disordered again. However, at this stage two hydrogen atoms of water molecule could be found from the difference Fourier synthesis. The two hydrogen atoms were refined isotropically with fixed positions and the three disordered oxygen atoms were left as they were because of lowest R1 value and the lowest difference peak value in Fourier difference synthesis. A bite large estimated standard deviations in all data might be due to vibrational and rotational disorder of the perchlorate group. The crystallographic data, conditions used for the intensity collection, and some features of the structure refinement are listed in Table 1.
Results and Discussion
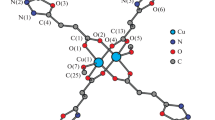
An ORTEP diagram [21] of 1 with the atomic numbering scheme is shown in Fig. 1. Selected bond lengths and angles are listed in Table 2. The monodeprotonated paba ligand acts a tridentate ligand. The copper atoms are bridged by a syn–anti carboxylate group of the paba ligand to form a polymeric one-dimensional chain. The intramolecular Cu···Cu separation is 9.370(3) Å, whereas the shortest intermolecular Cu···Cu one is 4.977(2) Å. The coordination environment of each copper(II) ion is best described as a square-plane with two nitrogen atoms, one carboxylate oxygen atom of the ligand, and one oxygen atom belonging to the carboxylate group of an adjacent molecule [Cu–N(1) 1.969(5) Å, Cu–N(2) 1.991(5) Å, Cu–O(1) 1.940(4) Å, Cu–O(2) 1.946(3) Å]. However, the Cu–Ow interatomic distance of 2.700(6) Å is well outside of any previously determined distances in related systems {[Cu(μ-papa)(H2O)](ClO4) · 2H2O} n [Hpapa = 2-(pyridylmethyl)amino-3-propionic acid; 2.310(4) Å] [14], {[Cu(μ-pmva)(H2O)](ClO4)}n [Hpmva = bis(2-pyridylmethyl)amino-5-valeric acid; 2.400(6) Å] [16], and {[Cu(μ-pmca)(H2O)](ClO4)} n [Hpmca = bis(2-pyridylmethyl)amino-6-caproic acid; 2.318(3) Å] [16]. The copper atom is displaced 0.051(2) Å from the least-squares plane defined by the N2O2 basal plane toward the water molecule. The water molecule forms hydrogen bond with the perchlorate oxygen atom O(4a) [Ow–HOw(1)···O(4a) 2.88(2) Å, 126.6(5)°]. The Cu–N(1) distance is comparable to that observed in {[Cu(μ-pmba)(H2O)](ClO4) · 3H2O} n [1.978(6) Å] [15] in which the copper(II) ion adopts a distorted square-pyramidal geometry. The N(1)–Cu–N(2) bite angle of the five-membered chelate ring is 83.7(2)°. The dihedral angle (α) between the plane of the carboxylate group and CuN2O2 plane is 40.6(2)°. Interestingly, the average N(2)–C [1.471(7) Å] distance involving the tertiary nitrogen is approximately 0.13 Å longer than the average N(1)–C [1.341(7) Å] distance involving the secondary nitrogen. This result may be attributed to the sp3 hybridization by protonation of the tertiary nitrogen atom.

An ORTEP view of {[Cu(μ-paba)](ClO4) · H2O} n (1) with the atomic numbering scheme (30% probability ellipsoids shown). The hydrogen atoms of carbon atoms are omitted for clarity
The IR spectrum of 1 reveals ν(NH) band at 3237 cm−1 associated with tertiary amine. The strong bands at 1489–1593 cm−1 are characteristic of the pyridine skeleton. The absorption band due to νas(COO) and νsym(COO) appears at 1615 and 1448 cm−1, respectively. The difference Δ = 167 cm−1, is suggesting a syn–anti bridging coordination mode for the carboxylate group [14], which is consistent with the crystal structure of 1. The visible absorption bands of 1 in the solid state and in solutions occur at 712 (solid), 715 (MeCN), and 714 nm (DMSO), characteristic of a copper(II) d zx ,d yz → d x2−y2 (2B1 → 2E) [22] transition in a tetragonal ligand field in which the copper(II) ion has a distorted square-pyramidal coordination environment. This fact may be due to the long Cu–Ow contact. Cyclic voltammogram for the copper(II) complex in 0.1 M TEAP–DMSO solution is shown in Fig. 2. Complex 1 exhibits two reversible one-electron waves corresponding to Cu(II)/Cu(III) and Cu(II)/Cu(I) processes. The oxidation and reduction potentials for 1 are +0.26 and −0.60 V vs. the Ag/AgCl reference electrode, respectively.

Cyclic voltammogram of {[Cu(μ-paba)](ClO4) · H2O} n (1) in 0.1 M TEAP–DMSO solution at 20.0 ± 0.1 °C. The scan rate is 100 mV/s
Supplementary Material
CCDC-290625 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at http://www.ccdc.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge, CB2, 1EZ, UK; fax: +44(0)1223-336033; e-mail: deposit@ccdc.cam.uk].
References
Pickart L, Goodwin WH, Burgua W, Murphy TB, Johnson DK (1983) Biochem Pharmacol 32:3868
Whitener GD, Hagadorn JR, Arnold J (1999) J Chem Soc, Dalton Trans 1249
Vigato PA, Tamburini S, Fenton DE (1990) Coord Chem Rev 106:25
Rardin RL, Tolman WB, Lippard SJ (1991) New J Chem 15:417
Tamboura FB, Gaye M, Sall AS, Barry AH, Jouini T (2002) Inorg Chem Commun 5:235
Seo JS, Whang D, Lee H, Jun SI, Oh J, Jin Y, Kim K (2000) Nature 404:982
Solomon DE, Sundaram UM, Machonkin TE (1999) Chem Rev 28:159
Jung OS, Pierpont CG (1994) J Am Chem Soc 116:2229
Evans OR, Xiong R, Wang Z, Wing GK, Lin W (1999) Angew Chem, Int Ed Engl 38:536
Kahn O (1993) Molecular magnetism. VCH publishers, Weinheim, Germany
Kahn O Martinez CJ (1998) Science 279:44
Floret F, Munno GD, Julve M, Cano J, Ruiz R, Caneschi A (1998) Angew Chem, Int Ed Engl 37:135
Han S, Manson JL, Kim J, Miller JS (2000) Inorg Chem 39:4182
Colacio E, Ghazi M, Kivekas R, Klinga M, Lloret F, Moreno JM (2000) Inorg Chem 39:2882
Choi K-Y, Jeon Y-M, Ryu H, Oh J-J, Lim H-H, Kim M-W (2004) Polyhedron 23:903
Choi K-Y, Park S-Y, Jeon Y-M, Ryu H (2005) Struct Chem 16:649
Choi K-Y, Jeon Y-M, Lee K-C, Choi S-N, Kim M-W, Lim H-H (2004) Trans Met Chem 29:405
North ACT, Phillips DC, Mathews FS (1968) Acta Crystallogr A 24:351
Sheldrick GM (1990) Acta Crystallogr A 46:467
Sheldrick GM (1997) SHELXL-97, Program for the refinement of crystal structures. University of Göttingen, Germany
Farrugia LJ (1997) J Appl Crystallogr 30:565
Hathaway BJ (1991) Struct Bonding, Berlin 57:2801
Acknowledgement
This study was financially supported by Kongju National University in the 2007 Star Project Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Choi, KY. Synthesis and Crystal Structure of One-Dimensional Carboxylate-Bridged Copper(II) Complex with 2-Pyridylmethylamino-3-butyric acid. J Chem Crystallogr 38, 53–56 (2008). https://doi.org/10.1007/s10870-007-9280-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-007-9280-z