
Crystal of [Fe(Phen)3]Cl(PHB).2(PHBH).7H2O (1) is triclinic, space group P-1 with a = 12.0388(11) Å, b = 15.5286(14) Å, c = 15.7794(14) Å, α = 89.759(2)°, β = 75.818(2)°, γ = 71.900(2)° and Z = 2, (phen = phenanthroline, PHBH = p-hydroxybenzoic acid, PHB = p-hydroxybenzoate anion). The phen in adjacent Fe(phen)3 2+ cations are π–π interacted forming offset face to face (OFF) motifs. Juxtaposition of four phen ligands from two cations encapsulate an R2 2(8) dimeric unit of H-bonded PHBH molecules within a centrosymmetric box froming a filled aryl box motif (FAB). Alternation of OFF and FAB motifs form {OFF⋯FAB}∞ strands. The Fe(phen)3 2+ cation engages its phen ligands in π–π and/or CH–π interactions with two crystalographically different PHBH molecules and one PHB anion. Seven water molecules and a chloride anion per iron(II) trisphenanthroline cation fill empty spaces in the structure forming a hydrophilic cluster. Extensive intermolecular H-bond interactions occur between water molecules, chloride anions, PHBH molecules, and PHB anions. Thermal analysis of (1) was done under N2(g). The TG, and dTG curves revealed the expected mass losses. All associated processes are endothermic as shown in the DSC curve.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metal chelates [M(LL)3]n+ (where LL is 1,10-phenanthroline (phen) or a modified phen ligand) are important species for developing new diagnostic and therapeutic agents that can recognize and cleave DNA.1 – 3 Variation in the ligand or the metal in these complexes has provided easy understanding of DNA-bonding and cleavage.4 – 6 Ruthenium compounds for example, have shown intercalating properties with DNA.6 , 7 Another interesting property of phen metal complexes is the catalytic role of ferriin-ferroin couple in oscillating chemical reactions between organic substrates (malonic acid) and bromate ions known as Belousov—Zhabotinskii reactions,8 which involves periodic changes in concentrations of substances in the reactions.9 – 16
Because of their inherent pocket structure, [M(LL)3]n+ complexes have the ability to form outer sphere complexes with anions,17 – 19 neutral molecules,20 or combination of them.21 The interaction of counter ions and inclusion molecules affect the lattice crystal structure of iron phen complexes: H-bonded iodide anions and water molecules in minor pockets occupy spaces between layers of [Fe(phen)3]2+ cations in [Fe(phen)3]I2.2H2O.22 Perchlorate anions occupy all pockets and form alternating sheets with [Fe(phen)3]2+ cations, while water molecules fill empty spaces.23 In the complex [Fe(phen)3]sac2.sacH.6H2O, a 3-D H-bond network is formed with saccharinate anions (sac), free saccharin molecule (sacH) and water molecules leaving channels where cations reside.21 The hydrophobic character of the pocket made by aromatic phen ligands causes these complexes to embrace one another,24 or attract organic molecules through van der Waals π–π interactions.25 The embracing iron phenanthroline cations form zigzag chains and layers in [Fe(phen)3]I2.2H2O where two of phen ligands intrude into major pockets of the adjacent host cation in a parallel way.22 Benzene ring of nitrobenzene replaces one of the two intruding phen ligands and occupies one of the major pockets of the chelate cation through π–π interaction in [Fe(phen)3]I2.2H2O.C6H5NO2.25
Recently there has been growing interest in the supramolecular motif of metal phenanthroline complexes caused by aryl interactions between planar phen in an offset face–to–face (OFF),24 edge–to–face (EF) primary motif,26 or P4AE (parallel four fold aryl embrace) secondary motif which consists of one OFF and two EF primary motifs.24 , 27 These interactions can be extended as net in one, two and three dimensions.24
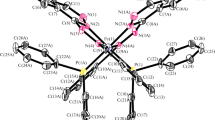
In this study we describe the supramolecular structure of (1) [Fe(Phen)3]Cl(PHB). 2(PHBH).7H2O, Fig. 1.

Thermal ellipsoids of the asymmetric part of the unit cell of (1) with 50% probability level.
Experimental
Physical measurements
Infrared data were collected on a Schimadzu 8300 FTIR spectrophotometer using KBr pellet method. The magnetic susceptibility of crystalline samples was measured on a Quantum Design Model 6000 Physical Property Measurement System. Elemental analysis of C, H, N were performed by Microanalytical Service, Department of Chemistry, University of Surrey, Guildford, Surrey, GU2 5XH. Thermal analysis was performed on Setaram-Labsys instrument using TG-DSC module. The heating rate was 2°C/min. Dry nitrogen was passed over 15 mg of the sample placed in a platinum crucible.
Synthesis of (1) [Fe(Phen)3]Cl(PHB).(PHBH)2.7H2O
To FeCl3.6H2O (0.54 g, 2.0 mmol) was added Phen (0.36 g, 2.0 mmol), and sodium p-hydroxbenzoate (0.64 g, 4.0 mmol) in 100 ml ethanol. The reaction was heated and then left to stand for one month, red platelet crystals of (1) was then formed in 25% yield. Anal. Calcd(%) for C57H55ClFeN6O16 (%): C, 58.39; H, 4.69; N, 7.17. Found: C, 57.15; H, 4.58; N, 7.25.
IR (KBr) cm−1: 3388.7 (w), 3116.7 (w), 3083.3 (w), 3018.8 (w), 2993.3 (w), 2817.8 (w), 2666.67 (w), 2547.8 (w), 1674.1 (s), 1608.5 (m), 1595.0 (m), 1510.2 (w), 1448.4 (m), 1425.3 (m), 1365.5 (w), 1317.3 (m), 1290.3 (m), 1244.0 (s), 1168.8 (m), 1128.3 (w), 1101.3 (w), 1012.6 (w), 929.6 (w), 854.4 (m), 769.5 (m), 723.3 (w), 692.4 (w), 640.3 (w), 619.1 (m), 547.7 (w), 503.4 (w), 422.4 (w).
Crystallographic data collection and solution of structure of (1)
Crystallographic data for (1) are summarized in Table 1. The crystal was mounted on a Bruker-AXS SMART APEX/CCD diffractometer using graphite monochromated MoKα (λ = 0.71073 Å ) radiation. Reflection data were measured at 100(2) K with a Φ and Ω scan mode. Intensities were corrected for Lorentz and polarization effects. The data were corrected for absorption using SADABS program.
The structure was solved by direct method using SHELXS-97. Nonhydrogen atoms were refined on F 2 anisotropically by full-matrix least squares method. The H atoms on carbon atoms were placed in calculated positions. Only part of the hydrogen atoms on oxygen atoms were found on difference electron density map. They were refined using AFIX 147 and AFIX 83 procedures. The final least-squares cycle gave R = 0.0780, wR2 = 0.1909 for 5678 reflections with I > 2σ(I) using the weighing scheme, w = 1/[σ2(Fo2) + (0.01000P)2 + 0.0000P] where P = (Fo2 + 2Fc2)/3.
Results
Description of the structure of the asymmetric unit of (1)
Complex (1) has been confirmed as diamagnetic low-spin by magnetic measurements. The asymmetric unit of (1) with atom labeling is shown in Fig. 1. Selected bond lengths (Å) and angles (°) are listed in Table 2. Complex (1) crystallized with one [Fe(phen)3]2+ cation, one chloride anion, one PHB anion, two PHBH and seven water molecules.
The iron(II) trisphen cation in (1) exhibits a slightly distorted octahedron geometry in which the iron atom is coordinated to six nitrogen atoms from three phen molecules with Fe-N bond distance ranging from 1.956(4) to 1.980(4) Å, Fig. 1, Table 2. The average 1.970(4) Å is in good agreement with the previously reported structures, 1.966(3) Å in [Fe (phen)3]sac2.sacH.6H2O,21 1.973(5) Å in [Fe(phen)3]I2.2H2O,22 1.978(3) Å in [Fe(phen)3](ClO4)2.0.5H2O,28 1.970 Å in L-[Fe(phen)3]bis[SbIII-D-tartrate].8H2O,29 and 1.980 Å in [Fe(phen)3]sac2.4H2O.30
Three phen ligands are bonded to iron forming five-member chelate rings. The bite angles between the three phen chelate rings and the iron atoms are similar: 82.81(16)°, 83.03(16)° and 82.70(16)° for angles N1–Fe–N2, N3–Fe–N4 and N5–Fe–N6 respectively, Table 2. The bite angle appears to correlate well with the metal-to-N-atom distance for different [M(phen)3]2+: 82.85(16)° and 1.970(4) Å in [Fe(phen)3]2+ in (1), 82.6(4)° and 1.978(3) Å in [Fe(phen)3]2+, 28 79.5(1)° and 2.097(2) Å in [Ni(phen)3]2+, 31 78.1(3)° and 2.127(14) Å in [Co(phen)3]2+, 32 79.25(5)° and 2.185(1) Å in [Cu(phen)3]2+, 33 70.2(2)° and 2.400(6) Å in [Hg(phen)3]2+ 34.
Accordingly, the regression analysis equation is represented by Y =−28.216 X + 138.741, where Y is the bite angle and X is the metal-to-N-atom bond distance, giving R = 0.972.
However, there are little deviation from linearity for the trans atoms with angles N1–Fe–N4, N3–Fe–N6, and N2–Fe–N5 at 176.96(15)°, 174.06(16)° and 174.81(16)° respectively owing to the rigidity of phen ligand, Table 2.35 The central Fe atom in (1) is close to coplanarity with each of the four surrounding N atoms, the deviations of Fe atom from the planes defined by the atoms N1, N2, N4 and N5; N1, N6, N3 and N4; and N2, N3, N5 and N6 are 0.071, 0.077, and 0.095 Å respectively. These values are close to those found in [Fe(phen)3](sac)2.sacH.6H2O,21 and [Fe(phen)3](sac)2.4H2O.37 Bond lengths and angles in the phen ligands of (1) are identical to those found in other structures.22 , 23 , 28 , 36
Trisphen Iron cations have three major pockets between the planar phen ligands and two minor pockets along the three-fold axis.17 Molecules or ions of suitable sizes can therefore intrude into these pockets and bind by electrostatic or dipole-dipole interaction to the [Fe(phen)3]n+ cations. However, the hydrophobic pockets made by aromatic phen ligands prefer inclusion of neutral molecules with π–ring systems over anions and water molecules.22 , 25 In compound (1), the aromatic PHBH molecules intrude into two major pockets and PHB anion into a minor pocket of the [Fe(phen)3]2+ cation, Fig. 1. In addition, seven water molecules and one chloride anion form a hydrophilic cluster on the outer sphere of the complex cation. The hydrogen atoms of water molecules are not refined, Fig. 1.
Hydrogen bonding network involving water molecules, p-hydroxybenzoic acid molecules, p-hydroxybenzoate and chloride anions
Seven water molecules with oxygen atoms O(3W), O(1W), O(5W), O(7W), O(6W), O(2W), and O(4W) are arranged in a close proximity to each other in an irregular chain forming a hydrophilic cluster, Fig. 1. The short separating distances between oxygen atoms of water molecules suggest the occurrence of H−bonds between them. The short contacts are: O(3W)⋯O (1W) = 3.095 Å, O(1W)⋯O(5W) = 2.957 Å, O(5W)⋯O(7W) = 2.702 Å, O(7W)⋯O (6W) = 2.587 Å, O(6W)⋯O(2W) = 2.903 Å, O(2W)⋯O(4W) = 2.855 Å . Water molecules in the cluster are also involved in H-bonding with other ions and molecules, Fig. 1. The H-bond distances (Å) and angles (°) are listed in Table 3:
-
i)
H–bond O(9)–H(9A)⋯O(3W). Water molecule with oxygen O(3W) acts as a H–bond acceptor from the hydroxide group O(9)–H(9A) of PHB anion.
-
ii)
H–bond O(1)–H(1A)⋯O(5W). O(1)–H(1A) from carboxylic group of PHBH (II) molecule acts as a H–bond donor to O(5W) of a water molecule.
-
iii)
H–bond O(6)–H(6A)⋯O(4W). Water molecule with oxygen O(4W) acts as a hydrogen bond acceptor from hydroxyl group O(6)–H(6A) of PHBH (I) molecule.
The chloride ion Cl(1) is also H–bonded to PHBH (II) molecule through its hydroxyl group O(3)–H(3), Fig. 1. The distances (Å) and angles (°) involved for O(3)–H(3)⋯Cl(1) are listed in Table 3.
Binding of p-hydroxybenzoic acid molecules and p-hydroxybenzoate anion with phen ligands in the pockets of [Fe(phen)3]2+ cation through π–π and CH–π interactions
An important feature in the outer sphere of complex (1) is the intrusion of two crystallographically different PHBH molecules into two major pockets and one PHB anion into a minor pocket of [Fe(phen)3]2+ cation, Fig. 1. PHBH molecules and PHB anions bind to phen ligands through π–π and CH–π interactions as shown in Figs. 2–5. These interactions disrupt the continuous interactions of phen in [Fe(phen)3]n+ cations in primary OFF motifs or secondary P4AE motifs and thus affect their expected mode of packing into 1, 2 or 3-D network observed in other compounds.24

p-hydroxybenzoic acid molecule PHBH (I) H-bonded to one another forming R 2 2(8) unit and involved in π–π interactions with phen ligands in (1).

Filled aryl box motif (FAB) made by juxtaposition of four phen ligands from two cations. The FAB is filled with H-bonded p-hydroxybenzoic acid PHBH (I) dimer.

CH–π interactions between phen ligands and benzoic acid molecule PHBH (II) in (1).

π–π interaction between PHB anion and phen ligand in (1).
The aromatic rings arrangement in (1) between PHBH (I) molecule or PHB anions with respect to phen ligands is a π–π stacked in an offset packing, Fig. 2 and 5.38 The short atoms contacts between aromatic rings (less than 3.8 Å) indicate the presence of medium strength π–π interactions in (1)39. The attractive van-der-Waals π–π interactions range from dipole–dipole to induced dipole-induced dipole electrostatic interactions; also present are the attractions between the positively charged hydrogen atoms and the π–electrons of the slipped rings. These attractive forces overcome the π–π Pauli repulsions caused by filled electron clouds of interacting rings.38 – 40 The presence of electronegative nitrogen in the phen ring of (1) withdraws electron density rendering it electron deficient. This strengthen its π–π interaction because of decrease in electron-electron repulsions.39 – 41
Intemolecular interaction involving p-hydroxybenzoic acid molecule PHBH(I)
Figure 2 indicates the occurrence of π–π interaction between PHBH (I) molecule and phen ligand. The PHBH molecules intrude into two major pockets of two opposing iron(II) trisphenanthroline cations. Each cation has two of its phen parallel to facing phen ligands of an opposing cation forming a hydrophobic filled aryl box (FAB) around a center of symmetry.42 , 43 The FAB encloses a H-bonded dimer of PHBH (I) molecules and can be described as {[Fe(phen)3]2+(PHBH)2[Fe(phen)3]2+}, Figs. 2 and 3.
Each Hydroxide group of a PHBH (I) molecule protrudes to the outside while the carboxylic group intrudes to the inside of the aryl box and interacts through H-bond with a carboxylic group of an opposing PHBH (I) molecule, Fig. 2. O(4)H(4) of one PHBH (I) molecule acts as a hydrogen-bond donor to O(5) of an opposing coplanar molecule. The distances (Å) and angle (°) for O(4)–H(4)⋯O(5) are listed in Table 3. Two hydrogen bond ineractions of this type form a centrosymmetric R 2 2(8) dimeric unit of PHBH (I) molecules, Fig. 2. This type of H–bond dimer was found the most stable compared to other possible arrangement of p-chlorobenzoic acid dimer such as parallel or umbrella complex. The calculated binding energy of H–bonded dimer of p-chlorobenzoic acid 21.3 Kcal/mol is one order of magnitude greater than the binding energy of the umbrella complex (2.6 kcal/mol).44 The angles between two phenyl rings or two planes of carboxylic groups of opposing H-bonded molecules are 180.00°. The rotation angle of phenyl ring relative to the carboxylic group in PHBH (I) molecule is ø rot = 10.18°. Each PHBH (I) molecule is π–π interacting with a phen ligand in the FAB. The centroid separation between two slipped rings is 4.449 Å. Atoms contacts are 3.647, 3.248, and 3.802 Å, Figs. 2 and 3.
Intermolecular interactions involving p-hydroxybenzoic acid molecule PHBH (II)
Figure 4 shows the occurrence of CH–π interactions between PHBH (II) molecules and phen ligands in two opposing major pockets of two symmetrically related Fe(phen)3]2+ cations through a center of inversion. CH–π interaction is a kind of hydrogen bond operating between a soft acid and a soft base system. Database surveys on transition metal complexes gave evidence of the CH–π hydrogen bond being an important factor in controlling the crystal packing and molecular structure of coordination and organometallic compound. A short intermolecular CH–π distances between aromatic ring (less than 3.0 Å) is an indication for the occurrence of CH–π hydrogen bond.45 – 51 The centroids separation between the T-shaped rings was less than 5.0 Å.52 The energy of CH–π bonds between benzene rings estimated by high-level ab initio calculation is −2.46 kcal.mol−1.53 This is in agreement with Montecarlo optimization,38 and the experimental value from a high-precision ionization measurement.54 In compound (1), short distances have been found between CH of PHBH(II)⋯π (phen): CH39⋯C29 (2.783 Å) and CH38⋯C31 (2.821 Å). The same PHBH(II) acts also as a H-bond acceptor from phen. The short contacts have been found between CH (phen) ⋯π of PHBH(II): CH32⋯C42 (2.832 Å), CH32⋯C37 (2.844 Å) and CH32⋯centroid (2.737 Å). The centroids –centroids separations between PHBH (II) and each of two phen involved in CH–π are 4.579 Å and 4.811 Å . The rotation angle of phenyl group relative to the –CO2H group in PHPH (II) is ø rot = 5.94 deg.
Intermolecular interactions involving p-hydroxybenzoate
Also involved in π–π interactions with phen ligands are PHB anions, Fig. 5. The centroid separation between the aryl rings is 4.131 Å. The atoms contacts are 3.216, 3.551, 3.797 and 3.951 Å. In a close contact and at a symmetrical position to one PHB is located another PHB anion which is also engaged in a similar π–π interaction though in a reversed direction. The oxygen atoms contact between two facing carboxylate groups is 3.141 Å. The rotation angle of the phenyl ring relative to carboxylate group in PHB anion is 17.86 deg. Also present is another weak hydrogen bond CH/O between CH10 (phen) and O7 (PHB). The distances are C10−H10⋯O7 = 3.217 A and H10⋯O7 = 2.436 Å. The angle <(DHA) is 141.56°.
Also from the data that we have about the carboxylic and carboxylate groups:

It is clear from above that the difference in bond lengths of C–O within the carboxylic group is much greater than the one found in the PHB anion. Also, the average distances for C−O bond in carboxylate (1.264 Å) is less than average single bond C–O (1.309 Å) and greater than average double bond C=O (1.238 Å) in carboxylic acid group of PHBH. This supports our assignment of PHB anion.
Offset face to face “OFF” motif formed by π–π interacted phen ligands of adjacent [Fe(phen)3]2+ cations
π–π stacking interaction governs the self assembly of a variety of complexes and interlocked molecular compounds.55 π–π interaction has been widely studied between π–donors rings (electron-rich) and π–accepting rings (electron-deficient).40 It has been also observed between similar aromatic rings.44 – 60 The phen ligands of adjacent [Fe(phen)3]2+ cations in (1) π–π interact in an offset packing, Fig. 6. The phen ligands belong to the family of polyaromatic rings molecules containing nitrogen heteroatom, which has been extensively reviewed for their π–π interactions.39 The presence of electronegative nitrogen in the ring strengthens this kind of interaction because of less electron repulsion. The hetero atom also induces a charge dipole in the rings causing a dipole-dipole attraction. The separation of 3.416 Å between phen rings in (1) falls in the usual range (3.3–3.8 Å), and is the result of strong π–π interactions.38 – 40 , 59 , 60 The atom contacts are 3.676, 3.479 and 3.687 Å, Fig. 6. This type of interaction between phen ligands causes complex (1) to adopt an OFF primary motif, Fig. 6. An OFF motif is an offset face – to – face overlap of two parallel aromatic ligands. Primary OFF motif rarely occurs as isolated motifs in the crystal lattice of metal phen complexes, but it does occur in (1). Complex (1) does not contain an edge – to – face interaction between phen ligands EF motif. Thus it can not adopt a P4AE motif (parallel fourfold phenyl embrace) which consists of one OFF and two EF motifs and requires embracing of four phen ligands two from each molecule as in [Cu(phen)2I]+I3 −.61 The usual distance between planes of pair of aryl rings participating in an OFF interaction is about 3.4 Å. This distance is 3.416 Å in (1). An OFF motif involves wide range of overlap of 1, 2, or 3 rings. The centroid - centroid distance between phen ligands (centroid of central C6 ring of phen) is 3.857 Å in (1).

Primary OFF motif in the crystal lattice of (1), π–π interaction between phen ligands.
This represents overlap of three rings since it is less than 4.0 Å according to a Cambridge Structural Database search (CSD).24 The interplanar angle between central C6 rings of phen is 0.6° (nearly zero). A centroid contact of 3.857 Å, a plane-plane distance of 3.416 Å and an angle of 21.68° between the ring normal and centroid vector, corresponds to a horizontal displacement of 1.358 Å from face to face alignment. This degree of offset between phen ligands lies in the expected range of 1–1.5 Å for three-ring overlaps. The usual intermolecular van der Waals attractive energy between two [M(phen)3]2+ cation is 9.8 Kcal/mol−1 with an OFF motif and 9.0 Kcal/mol with P4AE motif.24 , 61 Given that the contribution of coulombic repulsion is relatively negligible, both motifs are favorable thermodynamically to the same extent. However, complex (1) prefers OFF motif which involves only two phen ligands over P4AE motif which requires four phen ligands from the two interacting cations.

View of layer down crystallographic “a” axis: chains of (1) consist of alternating “OFF” and “FAB” motif enclosing PHBH(I) dimer. Other molecules and ions are omitted for clarity.
Supramolecular structure of (1)
The supramolecular structure of (1) is the result of self assemblies62 , 63 of Fe(phen)3]2+ cations and PHBH molecules into chains due to π–π interactions and H-bondings, Fig. 7.
Layer of propagating chains in (1) is observed in a view down crystallographic a axis, Fig. 7. Alternation of Fe(phen)3]2+ cations with “OFF motif” and PHBH (I) H-bonded dimeric unit in filled aryl box “FAB motif” forms the extended ⋯OFF⋯FAB⋯ OFF⋯FAB⋯chain, Fig. 7. Within the chain each PHBH (I) molecule engages in π–π interactions with phen of Fe(phen)3]2+. The separating distance between iron atoms of adjacent iron(II) trisphen cations engaged in “OFF motif” is 9.972 Å, while the separation between iron atoms in adjacent iron(II) trisphen cations engaged in “FAB motif” is 10.881 Å. PHB anions and PHBH molecules are aligned. Hydrophilic cluster of water molecules and chloride anions form extensive intra and inter H-bonding network which also involves PHBH molecules and PHB anions. The primary OFF motif interaction occurs only as isolated motifs in the crystal lattice of (1). This is different from the supramolecular structures of many other metal phen complexes which contain extended interaction of cations in an OFF motif and or P4AE motifs into 1, 2 and 3–D supramolecular structures such as OFF stack in [Cu(phen)(NO3)(H2O)2] NO3,24 OFF zigzag chains with outer form in [Fe(phen)2(NCS)2],64 , 65 or inner form in [Fe(phen)3](I3)2.S (S = CH2Cl2, acetone),43 as P4AE chains in [Fe(phen)3](I3)2.S (S = acetonitrile),43 and M(phen)3]I7 (M = Fe(II), Mn(II)),27 and as ⋯OFF⋯P4AE⋯chains in [Fe(phen)3](I3)2.S (S = H2O, toluene).43 The molecules of complex should participate in two motifs in order to form these OFF chains, P4AE chains or ⋯OFF⋯P4AE⋯chains.

Thermal analysis of complex (1).
A closely related to the “⋯OFF⋯ FAB⋯OFF⋯FAB⋯” chain structure in (1) is the [Fe(phen)3]I14 structure which consists of chains of alternating P4AE and FAB motifs: “⋯P4AE⋯FAB⋯P4AE⋯FAB⋯” where approximately linear I6 chain is threaded in the grooves of FAB.42 Other related structure is that of [Fe(phen)3](I3)2.CH3CN, where the layers contain chains of centrosymmetric P4AE of [Fe(phen)3]2+. FAB motifs occurring between chains encapsulate pairs of acetonitrile molecules [CH3CN⋯NCCH3].43 Similarly, the [M(phen)3]2+ cations in [M(phen)3]I7 (M = Fe, Mn) occur as parallel (P4AE)∞ chains. Segments of disordered polyiodides [I8]2− are positioned in the FAB motifs formed by (phen)2 grooves from complexes in adjacent (P4AE)∞ chains.27 Aryl box is also formed between chains of P4AE in the hydrophobic slab of [Co(phen)3][BF4]2.H2O.EtOH. This box is filled with two ethanol molecules with the ethyl group intruding to the inside and the hydroxyl group protruding to the outside of the box.24 The [Mn(phen)3]2+ cations in [Mn(phen)3](I3)2 form six fold aryl embrace 6AE . The structure contains FAB encapsulating I3 − anions.43 , 66 However, the inclusion of aromatic PHBH (I) molecule into the cavity of FAB in complex (1) is enhanced by π–π interaction with phen walls and allows the intrusion of carboxylic group into the middle of hydrophobic box. In (1), the metal phen cation prefers interaction with PHBH molecules and PHB anion rather than with the hydrophilic cluster. This reveals the ability of phen ligands to attract other neutral molecules with π–electrons systems. The interactions observed explain why [Fe(phen)3]2+ cation strongly salts in aromatic PHBH molecules and PHB anion in polar ethanol solvent. This is of great interest for the design of new important materials.
Thermal analysis of (1)
The thermal analysis data for complex (1) in the range 25°C–750°C under nitrogen atmosphere and heating rate of 2 °C/min are tabulated in Table 4.67 The DSC, TG and dTG curves are shown in Fig. 8. The % experimental mass variations are compared to % theoretical mass variations which correspond to expected molecular losses. Five solvated water molecules are first lost at 67.92°C then followed by two other water molecules at 124.81°C with an associated average endothermic heat of 13.32 J/g for each water molecule. Other losses are for phen and PHBH occurring in one step at 217.07°C, followed by phen and Cl− at 288.10°C, and PHBH at 367.27°C.
References
Barton, J.K. Science 1986, 233, 727.
Naing, K.; Takashani, M.; Tanigchi, M.; Yamagishi, A. Inorg. Chem. 1995, 34, 350.
Zelenko, O.; Gallagher, J.; Sigman, D.S. Angew. Chem. Int. Ed. Engl 1997, 36(24) 2776.
Sammes, P.C.; Yahioglu, G. Chem. Soc. Rev. 1994, 327.
Sigman, D.; Graham, D.R.; D’Aurora, V.; Stern, A.M. J. Biol. Chem. 1979, 254(12) 269.
Liu, F.; Wang, K.; Bai, G.; Zhang, Y.; Gao, L. Inorg. Chem. 2004, 43, 1799.
Mie, H.Y.; Barton, J.K. J. Am. Chem. Soc. 1986, 108, 7414.
Zhabotinskii, A.M.; Rovinskii, A.B. Theor. Exp. Chem. 1979, 15, 25.
Noyes, R.M. J. Am. Chem. Soc. 1980, 102, 4644.
Ganathisubramanian, N.; Noyes, R.M. J. Phys. Chem. 1982, 86, 5158.
Kovalenko, A.S.; Tikhonova, L.P.; Roizman, O.M.; Protopov, E.V. Theor. Exp. Chem. 1980, 16, 46.
Jwo, J.J.; Noyes, R.M. J. Am. Chem. Soc. 1975, 97, 5422.
Smoes, M.L. J. Chem. Phys. 1979, 79, 4669.
Shakhashiri, B.Z, Gordon, B.Z. J. Am. Chem. Soc. 1969, 91, 1103.
Singh, C.M.; Mishra, H.C.; Upadnyyay, R.N. J. Indian chem. Soc. 1979, 56, 835.
Yatsimirskii, K.B.; Tikhonova, L.P. Coord. Chem. Rev. 1985, 63, 241.
Beck, M.T. Coord. Chem. Rev. 1968, 3, 91
Johansson, L. Chem. Scr. 1976, 9, 30
Johansson, L. Chem. Scr. 1976, 10, 72
Johansson, L. Chem. Scr. 1976, 10, 149
Deng, R.M.K.; Simon, S.; Dillon, K.B.; Goeta, A.E. Acta Cryst. 2001, C57, 4.
Johansson, L.; Molund, M.; Oskarsson, A. Inorg. Chim. Acta 1978, 31, 117.
Baker, J.; Engelhardt, L.M.; Figgis, B.N.; White, A.H. J. C. S. Dalton 1975, 530.
Russell, V.; Scudder, M.; Dance, I. J. Chem. Soc., Dalton Trans. 2001, 789.
Fujiwara, T.; Iwamoto, E.; Yamamoto, Y. Inorg. Chem. 1984, 23, 115,
Dance, I.G. The Crystal as a Supramolecular Entity ed. G.R. Desiraju, John Willey, New York, 1996; pp 137–333.
Horn, C.; Scudder, M.L.; Dance, I.G. Cryst Eng Comm 2000, 2(36) 196.
Koh, L.L.; Xu, Y.; Hsieh, A.K.; Song, B.; Wu, F.; Ji, L. Acta Cryst. 1994, C50, 884.
Zalkin, A.; Templeton, D.H.; Ueki, T. Inorg. Chem. 1973, 12, 1641.
Li, J.; Wei, J.; Wu, X.; Zhu, N.; Zhang, Y. Cryst. Res. Technol. 1993, 28, 11.
Han, Q.; Jian, F.; Lu, L.; Yang, X.; Wang, X. J. Coord. Chem. 2002, 55(6) 633.
Boys, D.; Escobar, C.; Wittke, O. Acta Cryst. 1984, C40, 1359.
Anderson, P.O. J. Chem. Soc., Dalton Trans. 1973, 1237.
Deacon, G.B.; Raston, C.L.; Tunaley, D.; White, A.H. Aust. J. Chem. 1979, 32, 2195.
Goodwin, H.A.; Kepert, D.L.; Patrick, J.M.; Skelton, B.W.; White, A.H. Aust. J. Chem. 1984, 34, 1817.
Rund, J.V. Acta Cryst. 1980, B36, 3103.
Li, J.; Wei, J.; Wu, X.; Zhu, N.; Zhang, Y. Cryst. Res. Technol. 1993, 28, 11.
Jorgensen, W.L.; Severance, D.L. J. Am. Chem. Soc. 1990, 112, 4768.
Janiak, C. J. Chem. Soc., Dalton Trans. 2000, 3885.
Claessens, C.G.; Stoddart, J.F. J. Phys. Org. Chem. 1997, 10, 254.
Kabbani, A.T.; Zaworotko, M.J.; Abourahma, H.; Walsh, R.D.; Hammud, H.H. J. Chem. Crystallogr. 2004, 34(11) 749.
Horn, C.; Dance, I.G.; Scudder, M.I. Cryst Eng Comm. 2001, 3(2), 9.
Horn, C.; Berben, L.; Chow, H.; Scudder, M.I.; Dance, I.G. Cryst Eng Comm. 2002, 4(2), 7.
Sundholm, D.; Sundberg, M.R.; Uggla, R. J. Phys. Chem. A. 1998, 102(36), 7137.
Suezawa, H.; Hashimoto, T.; Tsuchinaga, K.; Yuzuri, T.; Sakakibara, K.; Hirota, M.; Nishio, M. J. Chem. Soc., Perkin Trans. 2000, 2, 1243.
Umezawa, Y.; Tsuboyama, S.; Honda, K.; Uzawa, J.; Nishio, M.; Bull. Chem. Soc. Jpn. 1998, 71, 1207.
Takahashi, O.; kohomo, Y.; Iwasaki, S.; Saito, K.; Iwaoka, M.; Tomoda, S.; Umezawa, Y.; Tsuboyama, S.; Nishio, M. Bull. Chem. Soc. Jpn. 2001, 74, 2421
Suezawa, H.; Yoshida, T.; Umezawa, Y.; Tsuboyama, S.; Nishio, M. Eur. J. Inorg. Chem. 2002, 3148.
Steiner, T.; Desiraju, G.R. Chem. Commun. 1998, 891.
Dubler, E.; Haring, U.K; Scheller, H.; Baltzer, P.; Sigel, H. Inorg. Chem. 1984, 23, 3785.
Bogdanovic, G.A.; Spasojevic-de Brie, A.; Zaric, S.D. Eur. J. Inorg. Chem. 2002, 1599.
Nishio, M. Cryst Eng Comm 2004, 6(27), 130.
Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. J. Am. Chem. Soc. 2002, 124, 104.
Grover, J.R.; Walters, E.A.; Hui, E.T. J. Phys. Chem. 1987 91, 3233.
Lehn, J.-M. Supramolecular Chemistry: Concept and Perspectives; VCH: Weinheim, 1995; pp 89–135.
Glidewell, C.; Howie, R.A.; Low, J.N.; Skakle, J.M.S.; Wardell, S.V.; Wardell, J.L. Acta Crystallogr. 2002, B58, 864.
Tong, M-L.; Li, W.; Chen, X-M.; Zheng, S-L.; Ng, S.W. Acta Crystallogr. 2002, C58, m232.
Garden, S.J.; Fontes, S.P.; Wardell, J.L.; Skakle, J.M.S.; Low, J.N.; Glidewell, C. Acta Crystallogr. 2002, B58, 701.
Hartshorn, C.M.; Steel, P. J. Inorg. Chem. 1996, 35, 6902.
Cozzi, F; Siegel, J.S. Pure Appl. Chem. 1995, 67, 683.
Horn, C.; Ali, B.F.; Dance, I.G.; Scudder, M.L.; Craig, D.C. Cryst Eng Comm. 2000, 2, 6.
Lindsey, J.S. New. J. Chem. 1991, 15, 153.
Braga, D.; Grepioni, F.; Desiraju, G.R. Chem. Rev. 1998, 98, 1375.
Konig, E.; Ritter, G.; Kulshreshia, S.K. Chem. Rev. 1985, 85, 219.
Konig, E. Prog. Inorg. Chem. 1987, 35, 527.
Kamalakshmi, D.; Reddy, K.R.N.; Padmavathy, D.; Rajasekharam, M.V.; Arulsamy, N.; Hodgson, D.J. Inorg. Chim. Acta 1999, 284, 158.
George, T.A.; Hammud, H.H.; Isber, S. Poyhedron 2006, 25, 2721.
Acknowledgment
Thanks to University of South Florida, Tampa for X-ray facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Material
CCDC-281055 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK.
Rights and permissions
About this article
Cite this article
Zaworotko, M.J., Hammud, H.H. & Kravtsov, V.C. The co-crystal of iron(II) complex hydrate with hydroxybenzoic acid: [Fe(Phen)3]Cl(p-hydroxybenzoate).2(p-hydroxybenzoic acid).7H2O. J Chem Crystallogr 37, 219–231 (2007). https://doi.org/10.1007/s10870-006-9174-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-006-9174-5