
Abstract
We have previously reported on the measurement of exact NOEs (eNOEs), which yield a wealth of additional information in comparison to conventional NOEs. We have used these eNOEs in a variety of applications, including calculating high-resolution structures of proteins and RNA molecules. The collection of eNOEs is challenging, however, due to the need to measure a NOESY buildup series consisting of typically four NOESY spectra with varying mixing times in a single measurement session. While the 2D version can be completed in a few days, a fully sampled 3D-NOESY buildup series can take 10 days or more to acquire. This can be both expensive as well as problematic in the case of samples that are not stable over such a long period of time. One potential method to significantly decrease the required measurement time of eNOEs is to use non-uniform sampling (NUS) to decrease the number of points measured in the indirect dimensions. The effect of NUS on the extremely tight distance restraints extracted from eNOEs may be very pronounced. Therefore, we investigated the fidelity of eNOEs measured from three test cases at decreasing NUS densities: the 18.4 kDa protein human Pin1, the 4.1 kDa WW domain of Pin1 (both in 3D), and a 4.6 kDa 14mer RNA UUCG tetraloop (2D). Our results show that NUS imparted negligible error on the eNOE distances derived from good quality data down to 10% sampling for all three cases, but there is a noticeable decrease in the eNOE yield that is dependent upon the underlying sparsity, and thus complexity, of the sample. For Pin1, this transition occurred at roughly 40% while for the WW domain and the UUCG tetraloop it occurred at lower NUS densities of 20% and 10%, respectively. We rationalized these numbers through reconstruction simulations under various conditions. The extent of this loss depends upon the number of scans taken as well as the number of peaks to be reconstructed. Based on these findings, we have created guidelines for choosing an optimal NUS density depending on the number of peaks needed to be reconstructed in the densest region of a 2D or 3D NOESY spectrum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The nuclear Overhauser enhancement, or NOE, is one of the most informative and widely used NMR observables for liquid-state NMR structure determination (Neuhaus and Williamson 2000; Wüthrich 1986). The conventional use for NOEs is as semi-quantitative upper limit distance restraints, the sheer number of which, along with other NMR restraints, can be used to converge a structure to its global minimum (Wüthrich 1986). This application, however, discards information including exact internuclear distances as well as dynamics, which are encoded in the cross-relaxation rate constants. There are a number of reasons why this information cannot be extracted reliably from conventional NOE measurements (Kumar et al. 1981), but the primary reasons are spin diffusion from neighboring atoms (Kumar et al. 1981; Kalk and Berendsen 1976; Keepers and James 1984) and dynamics (Brüschweiler et al. 1992; Post 1992; Bürgi et al. 2001; Zinovjev and Liepinsh 2013) as well as errors introduced from NOESY pulse sequences themselves (Strotz et al. 2015). We have previously developed a method (‘eNOE’ for exact NOE) (Orts et al. 2012; Vögeli 2014a) which takes into account some of these errors via the full-matrix relaxation formalism (Keepers and James 1984; Boelens et al. 1988, 1989) or a simplified formalism summing up the effects of all three-spin systems relevant for the eNOE under study, allowing for measured cross-relaxation rate constants to be converted into exact distances between protons (Vögeli 2009). The method for measuring eNOEs has been covered thoroughly in previously published reviews (Vögeli 2014a, b; Nichols 2017, 2018a). Using the eNOE protocol we have, among other applications, extracted distances up to 5 Å from GB3 and ubiquitin with only 0.1 Å error in the backbone (Vögeli 2009; Vögeli et al. 2010), calculated a high-resolution structure of a thermostable 14-mer UUCG RNA tetraloop (Nozinovic et al. 2009) using eNOEs alone (Nichols 2018b), and extracted relatively accurate distances between selectively labeled methyl groups within the 360 kDa proteasome from Thermoplasma acidophilium (Chi et al. 2018). In cases where robust eNOE networks can be measured, the averaged nature of the NOE enhancement allows for multi-state structures to be calculated that capture their spatial sampling (Vögeli et al. 2013, 2016). Using eNOEs we calculated a two-state ensemble of cyclophilin A, which uncovered a regulatory allosteric network between the enzyme’s active site and a nearby loop (Chi 2015a). Therefore, we have established that exact NOEs are a powerful tool that can be used to help investigate a diverse range of structural questions.
The measurement of eNOEs is simple, and only requires three additional NOESY spectra with different mixing times than would be normally required in the course of a structure determination project. However, this additional time requirement may still be a major discouraging factor. Measuring a 2D [1H-1H]-NOESY buildup series can be accomplished in a few days, as was the case for our work on 14-mer UUCG tetraloop. However, measurement of a full 3D buildup series on a 13C- and 15N-labeled protein can take up to 10 days or longer. As an example, we measured a 3D simultaneously [13C,15N]-resolved [1H-1H]-NOESY buildup series comprised of a total of five NOESY spectra on full-length H. sapiens Pin1, a 163-residue peptidyl-prolyl cis–trans isomerase (Lu et al. 1996), over a stretch of 10 days. This can be quite expensive on high-field NMR spectrometers, or even impossible to do on samples that are not stable over such a long measurement. Additionally, limits on measurement time will in turn limit the number of points and scans that can be acquired, putting a cap on the obtainable spectral resolution and requiring highly concentrated samples.
A technique which was first applied to NMR in the late 1980s, non-uniform sampling (NUS), has become popular among the biomolecular NMR community and can be used to drastically decrease the overall measurement time for the required eNOE buildup series. NUS is an acquisition method for NMR experiments containing time evolutions in indirect dimensions that only samples a subset of the full number of indirect points on the Nyquist grid (Barna et al. 1987; Hoch 1989; Schmieder et al. 1994; Hyberts et al. 2014). The missing points in NUS data necessitate alternatives to conventional Fourier reconstruction, and many successful approaches have been demonstrated, including iterative soft thresholding (Hyberts et al. 2012a) (IST), multidimensional decomposition (Hiller et al. 2009; Orekhov et al. 2003) (MDD), maximum entropy (Stern et al. 2002) (ME), compressed sensing (Holland et al. 2011) (CS), sparse multidimensional iterative lineshape-enhanced (Ying et al. 2017) (SMILE) reconstruction, and machine learning (Hansen 2019), with many of these methods integrated into common NMR processing frameworks such as NMRPipe (Delaglio 1995). In addition to NUS schemes which reduce measurement time, other NUS schemes and their reconstruction methods can be used to achieve signal-to-noise (SNR) increases for decaying data (Hyberts et al. 2013; Palmer 2015) and decreases in spectral artifacts due to signal leakage such as sinc-wiggles (Hyberts et al. 2012b), even at sampling percentages as low as 20% for 2D and 4% for 3D NMR experiments. NUS has been especially useful for work involving intrinsically disordered proteins, where resolution is the limiting factor, and NUS has enabled hyperdimensional NMR experiments to be acquired in an achievable measurement time (Jaravine et al. 2008).
However, many of these favorable attributes of NUS have only been identified for experiments with a low dynamic range, such as 3D backbone assignment experiments (Hyberts et al. 2014) and J-couplings (Born et al. 2018). For NMR experiments with a high dynamic range, such as NOESY experiments, decreasing NUS sampling percentages without complementary increases to the number of scans has been shown to cause a loss of weak cross peaks as well as the appearance of NUS-related spectral artifacts that can be mis-identified as peaks (Hyberts et al. 2009, 2012a, 2014). The structure of the gaps in the sampling scheme is a major determinant of the level of spectral artifacts, and it has been shown that Poisson gap schemes consistently produce spectra with high fidelity (Hyberts et al. 2010, 2012b) and have a low level of variance between random schedules (Hyberts et al. 2012b). Even for these schemes, the likelihood of generating subpar schedules using Poisson gap sampling is still relatively frequent (Aoto et al. 2014), illustrating the need for a robust schedule scoring method. While the present work uses only Poisson gap schemes, it should be noted that not all reconstruction methods perform best with the same type of NUS schedule (Ying et al. 2017; Hansen 2019), so that choice of an optimal sampling schedule type, even for data with little or no decay, is not always generalizable. Previous investigations of NUS applied to NOESY experiments have reported that sampling percentages below 30–40% cause significant deterioration (Hyberts et al. 2012b; Schlippenbach et al. 2018; Bostock et al. 2012). However, these findings were case specific and are also not generalizable because the NUS percentage required to successfully reconstruct a spectrum (relative to the fully sampled spectrum with the same parameters) depends upon the number and density of peaks in the data, and the necessity to capture sufficient signal to retain the smallest peaks of interest.
Thus, applying NUS to the eNOE protocol has the potential to substantially reduce measurement time, or when measurement time is not a limiting factor, could be used to obtain significantly better SNR or spectral resolution by acquiring additional scans and indirect points. However, it is important to determine what NUS percentage still produces quality eNOE data sets for a range of biomolecules of different molecular weight and proton density as well as to create guidelines for applying NUS to the eNOE method. To help make this determination, we took the fully sampled 3D NOESY buildup series consisting of five mixing times acquired from full-length Pin1 and the WW domain alone, as well as the 2D NOESY buildup series consisting of four mixing times acquired from a 14mer UUCG RNA tetraloop (Nozinovic et al. 2009), and resampled the data according to decreasing NUS sampling schemes in 10% increments generated using the Poisson gap method and reconstructed the resulting free induction decays (‘fids’) using SMILE (Ying et al. 2017). We then systematically investigated the quality of the extracted values of the auto- and cross-relaxation rate constants, the back-predicted diagonal peak intensities at zero mixing time, and the eNOE distances. Using simulations, we examined the impact of the number of spectral peaks on the reconstruction success. Using upper and lower limits for distance restraints, we calculated structures derived from the different NUS sampling percentages and compared them to those derived from the 100% sampling scheme. We then propose a general method and provide recommendations for the use of NUS for the eNOE method. While we focus on the application of NUS for the measurement of NOESY buildups, our findings are relevant to other applications of NUS. We note that the construction of the relationship between sample complexity and an optimal NUS density involves many variables to be considered and here we focus on a subset of these variables.
Results
The best possible outcome of applying NUS to a NOESY buildup series is to achieve the lowest possible measurement time without significantly sacrificing the quality of the data. Because reconstruction of NMR spectra using NUS depends on the number of signals to be reconstructed, we looked at three unique cases. The WW domain of Pin1 protein and the UUCG tetraloop RNA have roughly the same molecular weight (4.1 and 4.6 kDa for the WW domain and UUCG tetraloop, respectively). However, RNA molecules have a lower proton density compared to proteins and the 14mer UUCG tetraloop has good dispersion compared to larger RNAs. Thus, the WW domain is expected to require a slightly higher NUS density for successful reconstruction than the UUCG tetraloop. We also carried out our analysis with the most challenging system to which we have applied the eNOE method with the goal of calculating a multi-state structural ensemble, the 163-residue human Pin1. The full-length protein consists of two subdomains, the PPIase and WW domains, which have tumbling times of 14.1 and 11.3 ns respectively. Relevant parameters for the three samples are shown (Table 1).
We applied our previously established protocol for the extraction of eNOE-based upper- and lower-limit distance restraints (Orts et al. 2012; Vögeli 2014a). In short, four or more NOESY spectra with varying mixing times must be collected in a row. After assignment and removing any overlapped peaks, mono-exponential decay (diagonal peaks) and buildup curves (cross peaks) are fit over the increasing NOESY mixing times and used to derive auto- and cross-relaxation rate constants (ρ and σ). Fits of the diagonal peak decays are used to determine the initial magnetization at mixing time zero (M0), the value of which is then used to normalize the cross-peak buildups, and ρ. Simultaneously, corrections for spin diffusion calculated from a previously determined NMR or X-ray structure are applied to the measured cross-peak intensities. From these corrected intensities, the σ values are obtained via fitting with the previously obtained ρ values kept fixed. The output is a list of bi- (both symmetrically related cross peaks and their corresponding diagonals can be evaluated) and uni-directional (only one diagonal or cross peak can be evaluated due to overlap or low signal-to-noise) eNOEs, which are converted into inter-proton distances through the relationship σ ∝ r−6 (Solomon 1955). In our analysis, we extract the eNOE distances from σ values without considering the effects of internal dynamics. Thus, all motional effects are absorbed into the extracted distances referred to as effective distances reff (Vögeli 2014a).
In the following, we systematically investigate the quality of the values of ρ, σ, M0, eNOE distances (reff) obtained from five equally spaced mixing times with a maximum of 56 and 60 ms for full-length Pin1 and the WW domain, respectively, as well four equally spaced mixing times with a maximum of 160 ms for the UUCG tetraloop. After adding our standard tolerances to the reff to obtain upper- and lower-limit distance restraints (Strotz et al. 2017), we calculate structures derived from the different Poisson gap NUS sampling densities generated from the Wagner laboratory website and compare them to those derived from the 100% sampling case. We choose to analyze our results in terms of the NUS percentage of the fully sampled cases. Note that the same NUS percentage of different numbers of points that would be recorded for linear sampling may yield different results. Nevertheless, our results are general because we use numbers of points typically recorded for NOESY spectra.
Effect of decreasing NUS percentages on initial magnetization M0 and auto-relaxation rate constant ρ values
We first investigated the effect of decreasing NUS percentages on the quality of the fitted auto-relaxation rates ρ and the back-predicted initial magnetization values M0 of the diagonal-peak from the three samples. To do this, we made correlation plots of the M0 and ρ values for each of the decreasing NUS percentages versus the 100% sampling case (correlation plots are shown in Figs. S1, S2, and S3). While the quality of M0 was almost perfect even down to 10% sampling for all three cases (Figs. S1, S2, S3, top), an increasing number of outliers began to emerge in the plots of ρ as the sampling percentage was decreased (Figs. S1, S2, S3, bottom). The statistics for the correlation plots at decreasing NUS densities are shown (Fig. 1a for Pin1, Figs. S4a and S4b for the WW domain and UUCG tetraloop, respectively). The number of outliers was relatively large for Pin1 compared to the WW domain and UUCG tetraloop. For Pin1 the Pearson’s correlation coefficient R was reduced to 0.88 at 10% NUS, while for the WW domain and UUCG tetraloop, the correlation was barely impacted at all, with values of 0.98 and 0.99, respectively. The appearance of ρ outliers for Pin1 at decreasing NUS sampling densities was due to a loss of quality in a small subset of diagonal peaks (Fig. 1b). Out of the 466 analyzable diagonal peaks, only 10 of them experienced such issues, with the majority of the 10 only showing a reduction in quality below 40% sampling. This should not be considered an issue for the application of NUS to eNOE buildups because such outliers are easily identified by their sub-par decay plots (Fig. 1c) and subsequently removed from analysis (Orts et al. 2012; Vögeli et al. 2013). After excluding such outliers for all NUS densities used for Pin1, the resulting data set resulted in even higher quality correlations (Fig. S5 top and bottom, Fig. 1a, right). In addition, assuming that the cross peaks associated with excluded diagonals are still of reasonable quality, they can still be used in the form of generic normalized eNOEs (Chi et al. 2015b) (gn-eNOEs), thereby minimizing the overall loss of distance restraint information. Interestingly, the 10 outliers mentioned were equally split between Hα and Hβ protons, where all of the Hα protons were almost overlapped with the residual solvent signal and the Hβ protons were similar to the case shown in Fig. 1b, where the Hβ diagonal peak is poorly defined at the lowest sampling density. Because the resonance of Hα protons can be near to or overlapped with the residual solvent signal and are thus located in a region of high signal complexity, they will be affected by decreasing NUS densities overproportionally to the rest of the spectrum. Hβ protons, located in one of the most crowded regions of a protein spectrum, suffer from a similar issue. For the WW domain and UUCG tetraloop, errors to the ρ values began to appear at sampling densities of around 20%, although they were not significant enough to warrant exclusions. Pin1 represents an extreme case where the signal sparsity fraction was high enough in certain regions that some diagonal peaks were completely lost in addition to their cross peaks. This was in contrast to the WW domain and UUCG tetraloop where the signal sparsity was sufficiently small to avoid this effect at as low as 10% sampling. Overall, the correlations were of high quality indicating that, for the majority of diagonal peaks, decreasing NUS percentages has a minimal effect on the quality of the extracted M0 and ρ values down to 10% sampling. We also note that in contrast to M0, the extraction of the cross-relaxation rate constant from cross-peak buildups is relatively insensitive to the exact value of ρ. (Strotz et al. 2015).

Effect of decreasing NUS percentages on M0 and ρ values for Pin1. (a left) The slopes (m) and Pearson’s correlation coefficients (R) of the various correlation plots of M0 and ρ at each of the decreasing NUS percentages from Fig. S3 are graphed to show the overall trends. (a right) The same plot is shown except that M0 and ρ values from poor quality fits (from visual inspection of the decays) have been removed (raw plots are shown in Fig. S5). b The diagonal peak of the Hβ3 atom of residue 112 of Pin1 is shown for 100%, 40%, and 10% NUS sampling. The red cross hairs show the position of the peak assignment. c The fits of the diagonal peak decay of the Hβ3 atom of residue 112 of Pin1 are shown for 100%, 40%, and 10% NUS sampling. The x-axis shows the increasing NOESY buildup mixing times and the y-axis indicates the relative peak intensities
Effect of decreasing NUS percentages on cross-relaxation rate constant σ values and eNOE distances
We next investigated the effect of decreasing NUS sampling density on the quality of the fitted cross-relaxation rate constants σ and the corresponding eNOE distances reff. Because the intensity of cross peaks is very low compared to that of diagonal peaks, cross peaks are expected to suffer from decreasing NUS densities to a much larger extent. As before, we made correlation plots of the parameters of interest from each of the decreasing NUS percentages versus those at 100% sampling. Similar to the case with the ρ values, an increasing number of outliers emerged in the plots of the σ values and eNOE distances as the sampling density decreased (Figs. S6, S7, and S8). By 10% sampling, the Pearson’s correlation coefficient versus 100% sampling for σ and reff for Pin1 had decreased significantly to 0.61 and 0.88, respectively, indicating a significant loss in the quality of the peaks (Fig. 2a, left), as is visualized in Fig. 2b. For the WW domain and UUCG tetraloop the decrease in quality was not nearly as pronounced; at 10% NUS sampling density, the Pearson’s correlation coefficients for σ and reff decreased to 0.96 and 0.98 for the WW domain, and 0.99 and 0.95 for the UUCG tetraloop (examples of peak loss for the WW domain and UUCG tetraloop are shown in Figs. S9 and S10). We have previously introduced a measure for the quality of a buildup, χN, which quantifies the violation of the fit to the experimental intensities by taking the root-mean-square deviation over all mixing times (Vögeli et al. 2013). After visual inspection of the buildup curves and comparison with the spectra, we selected upper limit χN values of 27,500 for the WW domain, 29,000 for Pin1, and 35,000 for the UUCG tetraloop, above which eNOEs are automatically discarded, to be a sufficient filter of subpar buildups. Although these values vary due to the specificities of each studied case and are therefore somewhat subjective, they all agree well with values determined in our previous studies, which are all around 30,000. χN values above our selected cutoffs generally corresponded to cross peaks which were erroneous in the uniformly sampled case or no longer had sufficient signal-to-noise to be fit properly at lower NUS densities. In line with this, we binned the eNOE distances as a function of the χN value for the linear sampling cases which showed that the percentage of distances which violated the mean structure grew with increasing χN value (Fig. S11, left). In addition, following histograms of χN with decreasing NUS densities showed a count growth for all three cases past our selected cutoff (Fig. S11, right). An example of buildup curves from a bi-directional eNOE that was not affected and one in which only one of the two symmetrically related cross peaks was affected from the decreasing NUS sampling is shown (Fig. 2c).

Effect of decreasing NUS percentages on σ values and eNOE distances for Pin1. (a left) The slopes (m) and Pearson’s correlation coefficients (R) of the various correlation plots of the σ values and eNOE distances (reff) at each of the decreasing NUS percentages from Fig. S8 are graphed to show the overall trends. (a, middle) The same plot is shown except that buildups with χN values higher than the selected 29,000 have been discarded (raw plots for σ and reff are shown in Fig. S12 and Fig. 3). (a right) The slopes and Pearson’s correlation coefficients of the correlation plots of the bi- and uni-directional eNOE distances at each of the decreasing NUS percentages from Figs. S15 and S16 are shown. reff_bi-directional(i+j) means that the plot contains the distances calculated from each of the cross peaks which contribute to a single bi-directional eNOE. reff_bi-directional means that only the bi-directional distances are used. reff_uni-directional refers to distances solely from uni-directional eNOEs. b An example of cross peaks that decrease in quality with decreasing NUS percentages is shown. The symmetrically related cross peaks are shown within the inscribed boxes. c An example of a bi-directional eNOE which does not decline in quality with decreasing NUS sampling (top), and one in which only one of the cross peaks shows a well-defined decay (bottom), resulting in the conversion of the bi-directional eNOE into a uni-directional eNOE when the χN cutoff of 29,000 is applied
After removal of the χN violations, the resulting correlations for Pin1 (plots of σ and reff are shown in Figs. S12 and 3, respectively) were markedly improved with Pearson’s correlation coefficients for σ and reff increasing to 0.98 for both (Fig. 2a, middle). The same analysis is shown for the WW domain and UUCG tetraloop (Fig. S9c, right, and Fig. S10c, bottom, show the overall statistics while Figs. S13 and S14 show the correlation plots for the WW domain and UUCG tetraloop, respectively). These results indicate that once a suitable χN cutoff is selected, the quality of the eNOE distances does not deteriorate significantly even down to 10% sampling.

Correlation plots between eNOE distances from 100% sampling and decreasing NUS percentages for Pin1. Correlation plots between the calculated eNOE effective distances (reff) from the 100% sampling scheme on the x-axes of the plots and the specified NUS sampling percentages on the y-axes of the plots. The NUS percentages are indicated in the top left-hand corner of the correlation graphs. The slope (m) and Pearson’s correlation coefficient (R) of the plots are shown in the table located in the top left-hand corner of the figure. An upper limit χN value of 29,000 was selected after visual inspection of the buildup curves and all buildups which violated this number were discarded
Effect of decreasing NUS percentages on bi- and uni-directional eNOEs from Pin1
Because bi-directional eNOEs are calculated from the geometric average of the σ values from both of the symmetrically related cross peaks, we wondered whether the distances from bi-directional eNOEs would be more conserved than those from uni-directional eNOEs with decreasing NUS. For each NUS density, we looked at the correlation with 100% sampling of the uni-directional, both components of the bi-directional (distances from the cross peaks taken separately), and the averaged bi-directional eNOE distances (plots are shown in Figs. S15 and S16) from Pin1. While the correlations for all three data sets were superb (lowest R value of 0.97 for uni-directional reff at 10% sampling), they were slightly better for the bi-directional eNOEs, with the averaged bi-directional eNOEs having the highest Pearson’s correlation coefficient values (Fig. 2a, right). Indeed, the averaging of the two σ values into a bi-directional eNOE seems to absorb some of the error imparted by the decreasing NUS percentages (Fig. S15). It is also worth noting that the error imparted on the uni-directional eNOE distances from all decreased sampling percentages is less than the 20% error tolerance that would be given to them before being used as input for structure calculations (Strotz et al. 2015) (Fig. S16).
Effect of decreasing NUS percentages on eNOE yield and structure calculations
Due to the reduction in cross-peak quality with decreasing NUS densities, there was a significant loss in the overall eNOE yield after implementation of our χN cutoff values. Of the total 775 bi- and 2674 uni-directional eNOEs available at 100% sampling density for Pin1, only 371 bi- (48%) and 1761 uni-directional (66%) eNOEs remained at a 10% sampling level (Fig. 4a, bottom-left). The relative loss of bi-directional eNOEs was larger than for uni-directional eNOEs in part because of the conversion of many bi- into uni-directional eNOEs when only one cross peak of a pair was diminished in quality. We found that the number of eNOEs began to decline steeply for full length Pin1 at around 40% sampling, with 70% of the overall loss occurring from 30 to 10% NUS. For the WW domain, we observed a similar trend except that the NUS density at which the eNOE yield began to decline rapidly was about 30% NUS, instead of 40% as was the case for Pin1 (Fig. 5a, bottom-left). Out of the 264 bi- and 416 uni-directional eNOEs, 203 bi- (77%) and 330 uni-directional (79%) eNOEs remained by 10% sampling. For the UUCG tetraloop, the yield began to drop dramatically around 20% with 44 bi- (51%) and 170 uni-directonal (85%) eNOEs left at 10% out of the original 86 and 200 (Fig. 6a, right). As can be seen in Figs. 4b, 5b, and 6b, the eNOEs lost due to decreasing NUS densities make up primarily longer distance restraints and the percentage lost for each bin increases with reff. The loss of measured points in the indirect dimensions with decreasing NUS density results in decreased signal-to-noise of the spectrum causing a gradual loss of weak cross peaks. However, when the loss of weak peaks begins to increase abruptly as NUS sampling density is lowered, it indicates crossing over to the region of low probability of a successful reconstruction in the phase diagram theoretically derived by Monajemi and Donoho (2018) and experimentally refined in this work (vide infra), as there are no longer enough measured points to account sufficiently for the all the signals to be reconstructed.

Effect of decreasing NUS percentages on eNOE yield and structure calculations for Pin1. (a top-left) The CYANA target function (TF) and backbone bundle RMSDs of the WW and PPIase domains (from full-length Pin1) are plotted as a function of decreasing NUS percentages. (a top-right) The RMSDs between mean structures of the WW and PPIase domains of Pin1 from the 100% sampling scheme and at decreasing NUS percentages. Mean structures were created in Molmol (Koradi et al. 1996) and the RMSDs were calculated using CYANA. The horizontal lines show the RMSD of the bundle calculated from 100% sampling. (a bottom-left) The numbers of upper and lower limit distance restraints derived from eNOEs (including gn-eNOEs) are plotted as a function of the NUS percentage. These refer to the number of distance restraints before removal of distances which violate the structure by more than 0.6 Å (a bottom-right) The number of upper and lower limit distance restraints derived from eNOEs after the removal of distances which violated the structure by more than 0.6 Å. In all plots, the red error bars are the standard deviations calculated from 11 random 40% NUS schedules from the Wagner website and the blue points refer to the data from two NOESY buildup series measured with 20% and 10% sampling. The red dotted lines show when the eNOE yield decreases by 10% relative to the values at 100%. b The eNOE yield is binned by reff values for 100% (grey), 40% (orange), and 10% (red) sampling. c Superposition of the mean structures of the bundles calculated from each of the decreasing NUS data sets. The NUS percentages from which the structures are obtained are reflected in progressively fading colors, starting at black for 100% sampling

Effect of decreasing NUS percentages on eNOE yield and structure calculations for the WW domain. The same as in Fig. 4 with the WW domain

Effect of decreasing NUS percentages on eNOE yield and structure calculations for the UUCG tetraloop. The same as in Fig. 4 with the UUCG tetraloop
We used the eNOE data sets from each NUS percentage from the three cases to calculate structures in CYANA (Güntert et al. 1997; Güntert and Buchner 2015) and then recalculated structures after removing distances restraints violated by more than 0.6 Å. Normally, after confirming that the violated distances are not due to erroneous fits or spectral artifacts, we would include them in multi-state structure calculations because such distances report on spatial fluctuations (Vögeli et al. 2013, 2016). However, for the purposes of this paper, we are interested in the underlying quality of the data and such violations would skew the target functions in an unreliable manner. This eNOE trimming resulted in further loss of upper and lower distance restraints, but the overall trend with decreasing sampling percentages remained the same for all three cases (Figs. 4a, 6a, bottom right, 5a, bottom-left). For full-length Pin1, the structure backbone root-mean-square deviations (RMSDs) of the WW and PPIase domains gradually increased with decreasing sampling density (Fig. 4a, top-left). The WW domain and UUCG tetraloop structures, on the other hand, experienced minor RMSD (backbone RMSD for the WW domain and heavy-atom RMSD for the UUCG tetraloop) fluctuations around the linear sampling structure RMSD until 20% and 10% NUS for the WW domain and UUCG tetraloop, respectively, where they experienced relatively large jumps (Figs. 5a, 6a, top-left). These NUS densities are the points at which the eNOE yields begin to drop significantly as well, suggesting that the sudden increase in RMSD values is due to the large loss in total distance restraints. The RMSD values at low NUS densities for Pin1, the WW domain, and the UUCG tetraloop were still quite low, with maximal values of 0.74 and 0.51 for the PPIase and WW domain of full-length Pin1, 0.14 for the WW domain alone, and 0.62 for the UUCG tetraloop. In contrast, the CYANA target function (TF) decreased due to the loss of restraints, which contribute to the TF calculation.
To gauge how much the calculated structures lose accuracy relative to the 100% sampling case, we calculated RMSDs between the mean structures from the 100% sampling data sets and mean structures from the decreasing NUS sampling schemes. For Pin1, both domains showed a general increase in the backbone RMS deviation from the 100% mean structure with decreasing sampling density (Fig. 4a, top-right). However, the RMSD values for each NUS percentage were mostly smaller than their corresponding bundle RMSDs, indicating that divergence from the 100% NUS mean structure was driven by the overall loss of eNOEs and not by a decline in the quality of the distance restraint values. This trend was also true for the UUCG tetraloop (Fig. 6a, middle). In line with this, structural ensembles of Pin1 and the UUCG tetraloop comprised of mean structures from each NUS percentage had RMSD values similar to those from the 100% sampling case and showed that the structural variation between sampling schemes is mostly random (Figs. 4c, 6c). For the WW domain, the RMS deviation of all NUS mean structures from the full-sampling mean structure was larger than the RMSD of 100% NUS bundle. However, the WW domain RMSD was extraordinarily low with an RMSD of 0.03 Å and even at 10% NUS the RMSD from the 100% mean structure was only 0.34 Å (Fig. 5a, right). Again, a bundle composed of all of the mean structures shows little deviation (Fig. 5c).
In conclusion, even though a large portion of eNOE restraints is lost with low sampling percentages, the information content they carry is still sufficient to calculate high-resolution structures.
Finally, to confirm that the results of our analysis were consistent with eNOE data that is actually measured with NUS rather than resampled from a full linear sampling scheme, we acquired two eNOE buildup series with 20% and 10% sampling using the same schemes as before on Pin1. Pearson’s correlation coefficients of σ and reff versus 100% sampling were practically the same for both the reconstructed and measured data sets (compare Fig. S17 to Figs. 3 and S12). In addition, the structure statistics were also similar or even better than those for their corresponding structures from the reconstructed data sets (blue trend lines in Fig. 4). The minor differences between the reconstructed and measured data sets were likely due to the effectiveness of the solvent suppression along with slight degradation of the NMR sample.
Dependence on sampling schedule
While Poisson gap schedules have been shown to be fairly consistent in quality, there is still the potential to generate subpar NUS schemes (Hyberts et al. 2012b; Aoto et al. 2014). Because of this, we investigated the variability between 11 different random 40% Poisson gap schemes generated from the Wagner laboratory website on the Pin1 data set. Pin1 was chosen because it is the most affected by decreasing NUS densities and 40% because this is the cutoff before the spectral quality begins to decline rapidly. As with our previous analyses, we compared the values of interest from the different 40% sampling schemes to the 100% case. The σ and reff values across all 11 schemes were very consistent, with variations of the slopes of the σ and reff graphs of ~ 0.02 and 0.001, respectively, and Pearson’s correlation coefficients of ~ 0.99 for both (Fig. 7a). While schedule-specific outliers did appear over the different schedules, there were never more than a maximum of three per schedule (Fig. S18) and all of them violated the calculated structures by more than 0.6 Å and thus were removed from the final calculations. Variation in the structure statistics was also minimal (Fig. 7a, red error bars in Fig. 4), although there is a chance to generate schedules below the mean as was the case with schedule number 11 which had a lower overall eNOE yield. When we compared ensembles of mean structures of the WW and PPIase domains from each of the 40% NUS schedules, we found that the RMSD between all of the mean structures (0.48 and 0.54 Å for the WW and PPIase domains, respectively) were slightly above the average RMSD of each individual bundle (0.41 and 0.51 Å for the WW and PPIase domains, respectively). Visually the mean structures superimpose well (Fig. 7b). Importantly, this shows that the variations in the sampling schemes do not introduce a systematic error into the structure restraints. These results confirm that the variability between Poisson gap schemes is minimal. While NUS schemes can be optimized to account for overall correlation (τc) and relaxation times (Hyberts et al. 2012a), it is clear that for the most part, Poisson gap schemes should be sufficient to extract high-quality eNOE buildups.

Variation of eNOE parameters from 11 random 40% NUS schemes. a The plot on the left shows Pearson’s correlation coefficients and slopes for the correlation plots of σ and reff from the different 40% schedules versus the 100% sampling case. The four plots on the right are the same as in Fig. 4a, except that the eNOE yield and structure statistics are from 11 different 40% NUS schemes generated from the Wagner laboratory website, as indicated on the x-axis. b Similar to Fig. 4b, superpositions of the mean structures of the bundles calculated from each of the different 40% sampling schemes. The structures from the 11 40%-sampling schemes have different shades of grey, and the 100% mean structure is red. NUS schedule number 11 (marked with red asterisks) is the original 40% sampling scheme used in Fig. 4
Simulation of the dependence of the reconstruction success on the number peaks
The analysis of the three test molecules suggests that NUS percentage alone is not a good parameter to estimate the success in spectral reconstruction. It has been shown theoretically that whether a NUS scheme will reconstruct the fully sampled NMR spectrum successfully or not depends on whether the scheme samples enough of the k non-zero entries (here, parameter which define spectral peaks) of the sparse N-dimensional discrete signal, using n < N measurements, where n is the actually measured points and N the points of the 100% linear sampling case. (Monajemi and Donoho 2018) For a given NUS sampling density with the undersampling fraction δ = n/N, the probability of a successful recovery depends on the sparsity of the underlying signal, the sparsity fraction of which can be represented by ε = k/N. If a sufficient number of data points are measured such that the undersampling fraction δ is large enough compared to the sparsity fraction, then the probability of successfully reconstructing the spectrum will be close to 100%. This relationship gives rise to a so-called phase diagram (ε, δ) ∈ (0, 1)2 and a curve ε*asy(δ) which separates successful from failed reconstructions (Monajemi and Donoho 2018). In practice, an NMR spectrum is not uniformly sparse, rather, it has pockets of high density and low density, especially in the case of a NOESY spectrum. For example, the aliphatic region of proteins and ribose region of nucleic acids will be significantly denser than amide or imino regions. In the following, we use simulated reconstructions to investigate how the minimally required NUS sampling density is affected by the proton density of a sample, that is, the number of peaks in a spectrum.
There are two effects that can make spectra worse as NUS sampling density is reduced. One effect is reduction of signal, since if fewer increments are recorded, less signal is captured. While this reduction in signal can be somewhat mitigated by weighted sampling schemes (Palmer 2015), this is most helpful for systems where signals decay substantially in the indirect dimensions, which is not the case for typical biomolecular solution state measurements. The second effect can be thought of as a degrees-of-freedom requirement: the number of increments measured must be sufficient to describe the signals of interest.
To demonstrate the effect of the number of signals on NUS reconstruction quality, we used peak tables from the experimental data of Pin1 to prepare a series of simulated time-domain data sets with varying numbers of peaks, resampled these at several NUS densities, and performed reconstructions. In order to provide a more realistic test case, the time-domain simulations include random distortions in the form of small phase errors, unresolved couplings, and Gaussian noise. The reconstructions were performed via Iterative Soft Thresholding (IST) as implemented in NMRPipe (Hyberts et al. 2012a; Delaglio 1995; Stern and Hoch 2015), and IST extrapolation was not used (Stern et al. 2007; Hyberts et al. 2017). IST was used to avoid any possible bias in reconstructing simulated data via SMILE, which uses ideal time-domain signals to decompose the data. As an approximate way to gauge the quality of a given reconstruction, we compute the RMS difference between it and a reference “ideal” reconstruction. The ideal reconstructions were generated by simulating 3D time-domain data for one peak at a time, resampling the simulated single peak data according to a given NUS schedule, performing an IST reconstruction on the resampled single-peak data, and finally generating a complete spectrum by summing all the resulting single peak reconstructions. The single peak data includes random phase errors and unresolved couplings, but no Gaussian noise.
Simulations were generated with successively smaller subsets of peaks (specifically 378, 280, 181, 93, 39, and 18 peaks), and uniform random NUS sampling densities were varied from 5 to 100% in 5% steps, so that the combined results required roughly 7500 reconstructions. The results are summarized in Fig. 8 and example spectra are shown in Fig. S19. Figure 8a shows the agreement between a reconstruction and its ideal version for different numbers of peaks and at different NUS sampling densities. As shown, for a given number of peaks, agreement between a reconstruction and its ideal version is stable until a minimum NUS sampling density is reached, and below this sampling density, agreement gets worse quickly. This tallies with the results of Monajemi, who characterizes the success or failure of a reconstruction with respect to number of signals and NUS sampling density as an abrupt phase transition. (Monajemi 2016) In our example, the 378 peak case begins to get worse at 40% sampling density, while the 18 peak case still yields good results at 5% sampling density.

Impact of peak number on reconstruction success of chosen NUS density. a Relative RMS difference between IST reconstructions and their “ideal” counterparts for simulated data with various numbers of peaks at different NUS sampling densities. As shown, spectra with more peaks require higher sampling densities for a good reconstruction. b Same RMS difference as shown in (a), but displayed in terms of the ratio of NUS samples (measured points) to peak count (1/ε). It can be seen that all spectra, regardless of peak count, need about the same ratio of samples to peaks for a good reconstruction. This means that NUS sampling density must be chosen to provide enough total measured points for the expected number of peaks
As noted, degrees-of-freedom considerations require that the total number of NUS measured points must be sufficient to describe all of the signals in the data. The fully-sampled data has 8,000 increments that are complex in the two indirect dimensions, so that there are 32,000 total measured points. If we assume that each peak in an indirect plane encodes at least 5 parameters (two positions, two widths, height), then we can view the reconstruction with 378 peaks in an indirect plane as similar to extracting 378 × 5 = 1890 parameters k from 32,000 points N, a ratio of ~ 17 measured points per parameter (corresponding to 1/ε). We can expect that for a successful reconstruction, this ratio must be greater than one. In practice, to accommodate non-ideal signal shapes and random noise, and to account for the fact that a spectrum has to have more empty space than signal in order to identify peaks, we can anticipate that this ratio should be several times greater than one. This is demonstrated in Fig. 8b, which displays the results in terms of the ratio of total NUS samples to number of hypothetical peak parameters. As shown, all spectra for this example, regardless of the total number of peaks, give stable reconstructions up to a ratio of 10 measured points per parameter, and get worse quickly at lower ratios. This demonstrates clearly that the NUS sampling density must be selected so that the total number of increments measured is large enough to accommodate the maximum number of signals found in the indirect dimensions. This means that the minimum required sampling density for a 2D case will be determined according to the indirect column (sometimes referred to as “NOESY tower”) with the most signals, and for a 3D case it will be determined according to the indirect plane with the most signals. This point is further emphasized in Fig. S19, which shows reconstructions with 378, 181, and 18 peaks performed at 50%, 35%, 20% and 5% sampling density. As expected, all cases show decreasing signal to noise with lower sampling density. However, at 20% density, the 378 peak case shows many more visible reconstruction artifacts than the 181 peak case, while the 18 peak case is still mostly artifact free. At 5% density, the 378 peak case and 181 peak case have so many artifacts as to be unrecognizable, while the 18 peak spectrum retains its original arrangement of peaks, but at lower signal to noise.
The simulated reconstruction artifacts were also observed in our measured data as the emergence of what we describe as a “noise step” at low NUS densities. For Pin1 and the WW domain, it occurs when transitioning from 5 to 4.5 ppm in the direct 1H dimension of the NOESY spectra and there appears to be a second step that brings the noise back to the original level at ca. 0.5 ppm. The noise step continues increasing in severity with lower sampling percentages and is more significant for Pin1 than for the WW domain (Fig. S20). For the UUCG tetraloop, there was not one step but several corresponding to the aromatic and ribose regions of the NOESY spectra (Fig. S21). Interestingly, these noise steps are not uniform over the spectra but appear at discrete intervals corresponding to regions of high peak density. For Pin1 and the WW domain, they are located in the same region as the aliphatic carbon signals and are more dispersed for the WW domain presumably because of increased peak density in Pin1. For an RNA molecule, the regions of high peak density correspond to the ribose and aromatic regions, where there is often heavy peak overlap. In addition, these steps appear at similar NUS densities at which the eNOE yield and spectral quality begin to deteriorate rapidly, suggesting that they directly reflect the NUS density at which the reconstructions move from the “successful” region into the “failed” region of the phase diagram (Monajemi and Donoho 2018). They fail for the regions of high peak density first because these regions require a higher sampling density to be successfully reconstructed than the regions of lower peak density.
Discussion
We have conducted a detailed analysis of the quality of eNOE parameters extracted from simulated NUS schemes in 10% increments down to 10% sampling density for Pin1, the WW domain, and the UUCG tetraloop. Although the eNOE distances are still of good quality down to 10% sampling, the spectral quality and thus eNOE yield decay at NUS densities that depend upon the number of peaks needed to be reconstructed in the densest region of the NOESY spectrum. For Pin1 and WW domain data both recorded with 3D NOESY, this density was 40% and 20%, respectively, while for the UUCG tetraloop it was 10% in 2D NOESY. In the following, we create guidelines for what NUS density should be measured depending on how many total indirect points are to be collected as well as the maximum number of peaks within the highest density region of the spectra. However, as we mentioned before, these guidelines should be followed loosely because the optimal NUS density required will vary from case to case.
A guideline for choosing the optimal NUS density
While determining the exact NUS percentage required to successfully reconstruct an NOESY spectrum remains difficult to predict, our data has allowed us to devise guidelines that can be used in an approximate manner to estimate what the NUS % should be used for a particular sample. To do this, we calculated ε, where ε = k/N and k is the degrees of freedom associated with a peak (5 for 3D and 3 for 2D) and N is the number of points from the uniformly-sampled case, for Pin1, the WW domain, and the UUCG loop and plotted them versus the NUS % at which the NOESY data started to deviate from the uniformly-sampled case by more than 10% (Fig. 9). Pin1 and the WW domain had ε values of 0.03 (200 peaks and 32,000 points) and 0.006 (40 peaks and 32,000 points) and deviated by more than 10% from the uniformly-sampled case at 40% and 20%, respectively. These observations were in good agreement with the simulations from Fig. 8, which showed that 3D NOESY spectra containing a similar number of peaks began to deviate around the same NUS percentage. Fitting the simulated and measured values to a simple power function results in plots that show the “phase transition” between sampling space that is likely to result in successful and unsuccessful reconstructions of the spectra (Fig. 9a). The curve fit from the simulated points lies slightly above that from the measured, likely reflecting the additional imperfections inherent to a real NMR measurement which negatively impact the ability to successfully reconstruct a spectrum. Therefore, the curve fit from the measured data represents more practical guidelines when selecting a NUS percentage to use for a sample with a specific ε value.

NUS % recommendations for 3D and 2D NOESY. The plots show the ratios of number of signals to linear sampling points ε versus minimal NUS % required for successful reconstruction. a ε for 3D NOESY (k/N or 5 × the number of peaks in the indirect dimensions divided by the number of points, 32,000 in this case) is on the y-axis and the NUS % at which the spectra begins to deviate significantly from the uniformly-sampled case on the x-axis is plotted for both the values simulated in Fig. 8 and for the measured values from Pin1 and the WW domain. On the right side of the curve, a successful reconstruction of the 3D NOESY is predicted while a value that falls on the left side would be predicted to produce a subpar reconstruction. The grey area between the simulated and measured curves shows the deviation of the simulations from the experiment values. b ε for 2D NOESY (3 × the number of peaks on the most crowded 1D slice through the indirect dimension divided by the number of points, 400 for the UUCG tetraloop) and the NUS % at which the spectra deviated from the 100% NUS case
3D and 2D spectra are different in nature and vary dramatically in the number of points collected for a dataset and thus are not directly comparable. Therefore, we generated a second phase transition plot for 2D NOESY using the same power function as used for 3D, and calibrated it with the ε value obtained from the UUCG tetraloop (Fig. 9b). 20 peaks and 400 points resulted in an ε value of 0.15. The NOESY data started to deviate by more than 10% from the uniformly-sampled case at ca. 10% NUS. This was also in agreement with the simulations from Fig. 8b.
Protocol for measuring eNOE buildups with NUS
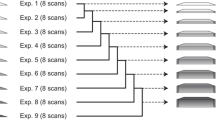
Based on our findings we recommend the following protocol for using NUS in measuring eNOE buildups (Fig. 10). First, a NOESY spectrum with a longer mixing time than for the buildups should be acquired with uniform sampling for assignment purposes. This guarantees that any NUS-related spectral artifacts that could be identified as peaks are not assigned. In addition, comparing the assigned NOESY to the longer mixing time spectra from the buildup series will give an estimate on how many peaks are lost due to the NUS. From the uniformly sampled NOESY, a count of the number of non-noise peaks in the most crowded region of the spectra should be obtained. Take this number and multiply it by five for 3D and three for 2D NOESYs and divide the resulting number by the number of points from the uniformly-sampled case to obtain ε (if the spectra has 100 by 50 complex points in the indirect dimensions, then the total number of points would be 2 × 100 × 2 × 50 = 20,000). Compare the calculated ε to the plots in Fig. 9 to determine the required NUS percentage. Next, a NOESY buildup series should be acquired using NUS with at least 4 different mixing times (note that the same NUS scheme needs to be used for each mixing point). The mixing times should also be kept low enough so that spin diffusion is moderate. Assuming an inverse relationship between the ideal maximum of the mixing time and the overall rotational correlation time (τc), we have derived theoretical optimal upper limit mixing times of approximately 2.5 × 10–10 and 4 × 10–10 s2τc−1 for proteins and RNA, respectively. (Vögeli 2014a; Nichols 2018b) After acquiring the NOESY buildup series, the assignment from the uniformly sampled NOESY should be transferred to the buildup series spectra with the longest mixing time, and then the raw peak intensities across all mixing times need to be extracted. We find that the NlinLS autofit script within NMRPipe (Delaglio 1995) provides a robust way to do this. In order to correct for spin diffusion, an existing 3D structure of the biomolecule of interest must be available. This can be a previously calculated NMR ensemble or X-ray structure. If no such structure exists, an initial structure can be calculated from the NOE data collected in the linearly sampled NOESY spectrum with the long mixing time using the conventional NOE approach. Next, the ρ and σ rates are fitted using either the eNORA2 Matlab version or the version integrated into CYANA. The fits of the diagonal peak decays are then inspected, and any subpar fits are removed. The M0 and ρ values obtained from these fits are used as fixed parameters when the σ are obtained from fits to the NOE buildups. The buildup fits are inspected and an upper limit χN quality cutoff is determined above which eNOEs will be removed from the data set. The output upper and lower limit distance restraints may then be used as input for structure calculations. If generic normalized NOEs (gn-eNOEs) (Chi et al. 2015b) derived from buildups with unresolved diagonals are desired, upper limit M0 and average ρ values must be used in place of the missing diagonals. The resulting upper limit distance restraints should be given an error tolerance of 20%.

eNOE protocol for NUS. The steps for applying NUS for the measurement of eNOEs is outlined. (1) After sample preparation, a NOESY spectrum with a longer mixing time is acquired with uniform sampling for the purpose of assignment. (2) Calculate ε for the NOESY spectra by multiplying the number of peaks in the indirect dimensions by five for 3D or three for 2D (number of peaks in the most crowded indirect plane or along the most crowded 1D slice, respectively) and divide by the number of points (indirect real points). Use this value to determine the NUS % required by Fig. 9. (3) Next, a NOESY buildup series with at least 4 mixing times is acquired with NUS sampling in order to reduce the measurement time. We recommend staying above 40% sampling. The optimal upper limit mixing times are approximately 2.5 × 10–10 and 4 × 10–10 s2τc−1 for proteins and RNA, respectively, where τc is the overall rotational correlation time (Vögeli 2014a; Nichols 2018b). (4) The assignment of the uniformly sampled NOESY is then transferred to the buildup series measured using NUS and (5) the buildups fit using eNORA2 (Matlab or CYANA version). If a 3D NMR or X-ray structure is available, it should be used as input for spin-diffusion corrections. Otherwise a conventional NMR structure has to be calculated first. (6) The diagonal peak fits are inspected visually, any erroneous fits removed, and an upper limit χN cutoff is selected for the cross-peak buildup fits above which fitted eNOEs are not converted into distance restraints. If gn-eNOEs are derived, upper limit M0 and average ρ values are used as input for the missing diagonal peaks, and the resulting upper limit distance restraints given a 20% tolerance. (7) Structures can then be calculated using the list of upper and lower limit distance restraints and the resulting structure can be used as input for spin diffusion corrections in an iterative process (8)
Dependence of eNOE fidelity on sample concentration, overall tumbling time, number of scans and points, and signal dispersion
In order for successful reconstruction of an NMR spectrum a sampling density must be chosen that samples enough points for the most complex region of a given spectrum. In addition to the number of peaks, there are a host of other parameters that may impact the quality of a NUS reconstruction to different extents. These include size and proton density of the biomolecule, whether it is folded or disordered, the concentration and number of scans, the spectrometer sensitivity and field, relaxation and line width depending on the rotational correlation time (τcorr) and internal dynamics, the generated schedule used to sample points, and likely many others. The effects of these parameters on NMR spectral quality are known. However, it is not clear to what degree increases to some parameters, such as sample concentration or the number of scans, would affect the resulting NUS reconstructions. Having a more highly concentrated sample may allow a lower NUS density to be used due to the increased signal-to-noise or it may have the opposite effect because there will be more peaks to be reconstructed. Similarly, an intrinsically disordered protein (IDP) may require a lower sampling density than for a folded protein because there would be significantly less cross-peaks. At the same time, however, IDPs are heavily overlapped and therefore would require a higher sampling density in order to have a sufficient number of points to successfully reconstruct such crowded regions. Our analysis here consisted of well-folded biomolecules with relatively dispersed spectra. Clearly, much further work is required in order to determine how such parameters affect the success of NUS.
Conclusion
We have investigated how NUS Poisson gap schemes with decreasing densities affect the quality of eNOE buildups and have determined that while many distance restraints are lost due to decreasing signal-to-noise, the majority of the remaining distances retain their quality. In addition to a gradual loss of cross peaks, there are NUS densities at which the spectra begin to deteriorate rapidly due to dropping below the minimal sampling density required to successfully reconstruct the spectrum. This density depends upon the complexity of the sample and thus the number of peaks needed to be reconstructed in the densest region of the NOESY spectra. For Pin1, this transition occurred at 40% sampling of 3D NOESY, while for the WW domain and UUCG tetraloop it occurred at 20% (3D) and 10% (2D), respectively. Using this data, we created loose guidelines for choosing an optimal NUS density for the measurement of eNOE buildups and have proposed a protocol for applying NUS to the eNOE method. The use of NUS will allow the measurement of eNOE buildups in a fraction of the normal time, cutting down the measurement time of a typical 3D NOESY series from ca. ten days to four or less. Although not investigated in the current work, the required time may be further reduced when the parameters describing a peak other than the intensity are determined from the uniformly sampled spectra and then fixed in a combined reconstruction and analysis of the NUS spectra.
Methods
Measurement and processing of uniformly sampled NOESY buildup series on Pin1
The recombinant expression of full-length Pin1 protein and purification, along with NMR experiments carried out for assignment have been described previously (Born 2018). The chemical shifts have been deposited in the BMRB (code: 27579). The 15N,13C-isotopically enriched Pin1 sample was 2 mM in 20 mM sodium phosphate, 50 mM sodium chloride, 5 mM dithiothreitol, 0.03% sodium azide, and 3% D2O at a pH of 6.5. The NOE buildup series was measured with 3D simultaneously [13C,15N]-resolved [1H-1H-X13C,15N] NOESY schemes (Vögeli et al. 2013) with incremented mixing times tmix of 56, 48, 40, 32, and 24 ms on a Varian 900 MHz spectrometer equipped with a cryogenic probe at 298 K. The spectra were recorded using a linear sampling scheme with 160(1H, t1) × 50(13C/15N, t2) × 1024(1H, t3) complex points, maximal evolution times of t1max,1H = 11.4 ms, t2max,15N/13C = 16.1 ms, and t3max,1H = 72.9 ms, spectral widths SW1,1H = 15.6 ppm, SW2,15N = 34 ppm, SW2,13C = 30 ppm, SW3,1H = 15.6 ppm, an interscan delay of 1.2 s, and 4 scans per increment resulting in a measurement time of ~ 2 days per spectrum. All spectra were processed with the NMRPipe/NMRDraw/NlinLS package (Delaglio 1995). The time-domain data were multiplied with a squared cosine function in the direct dimension and cosine functions in the indirect dimensions and the number of complex points were doubled by zero-filling once. A polynomial function was used for solvent suppression.
The measurement of the 20% and 10% NUS 3D NOESY buildups on Pin1 were acquired in the same way as for the uniform case except that the points in the indirect dimensions were acquired according to either the 20% or 10% Poisson gap NUS sampling schedules generated from the schedule generator 3.0 from the Gerhard Wagner laboratory website (https://gwagner.med.harvard.edu/intranet/hmsIST/gensched_new.html). For the 20% case, 1594 of the 8000 normally measured complex points on the Nyquist grid were acquired, and the measurement time was ~ 8.6 h per spectrum. For the 10% case, 807 of the 8000 normally measured complex points were acquired, resulting in a measurement time of ~ 4.3 h per spectrum. The fids were then processed and reconstructed in NMRPipe using SMILE.
Re-sampling of uniformly sampled data to NUS schedules and data processing for Pin1
Poisson gap sampling schedules were generated using the schedule generator version 3.0 from the Gerhard Wagner laboratory website. For all percentages (90% through 10% in steps of 10%), the number of complex points in the indirect 1H and 15N/13C dimensions was 160 and 50, respectively. The sinusoidal weight for all schedules was 2 and the seed value was 0 (random seed). The fid files from the uniformly sampled buildup series were then resampled according to the decreasing NUS sampling densities using the NUS processing tools available within NMRPipe. Specifically, the nusCompress.tcl script was used to delete complex points from the fully sampled fids that were not “measured” according to the given NUS schedules. The compressed fids were then processed following the conventional NUS protocol in NMRPipe. The nusExpand.tcl script was used to sort and expand the data so that the missing points were filled with zeros. The data were Fourier transformed as normal, and then reconstructed using the SMILE algorithim (Ying et al. 2017). This process was carried out for all NUS percentages.
Measurement and processing of uniformly sampled NOESY buildup series on the WW domain of Pin1
The sample preparation, NMR measurements, and data processing for the WW domain of Pin1 have been reported previousally (Strotz et al. 2015). The 15N,13C-isotopically enriched Pin1 sample was 1.2 mM in 10 mM potassium phosphate, 100 mM sodium chloride, 0.02% sodium azide, and 3% D2O at a pH of 6.0. The NOE buildups were measured using the same pulse sequence as for Pin1 with mixing times of 20, 30, 40, 50, and 60 ms. The spectra were acquired using a linear sampling scheme with 200(1H, t1) × 40(13C/15N, t2) × 1024(1H, t3) complex points, maximal evolution times of t1max,1H = 22.0 ms, t2max,13C = 7.6 ms, t2max,15N = 14.4 ms, and t3max,1H = 102.4 ms, spectral widths of SW1,1H = 13.0 ppm, SW2,13C = 29.8 ppm, SW2,15N = 39.2 ppm, and SW3,1H = 14.3 ppm, an interscan delay of 0.8 s and 4 scans per increment resulting in a measurement time of ~ 1 day per spectrum.
Re-sampling of uniformly sampled data to NUS schedules and data processing for the WW domain
The uniformly sampled data from the WW domain was resampled according to Poisson gap schedules (90% through 10%) and the data reconstructed in the same manner as for Pin1 except with 200(t1) × 40(t2) complex points for the indirect dimensions.
Measurement and processing of uniformly sampled NOESY buildup series on the 14mer RNA UUCG tetraloop
The sample preparation, NMR measurements, and data processing for the 14mer RNA UUCG tetraloop have been reported previously (Nichols 2018b). The unlabeled sample in H2O was 1 mM in 20 mM sodium phosphate, 0.4 mM EDTA, and 5% D2O. The NOE buildups for the sample in H2O were measured using a 2D WaterGate [1H-1H] NOESY with four mixing times (40, 80, 120, and 160 ms). The spectra were acquired using a linear sampling scheme with 200(t1) × 1470(t2) complex points, maximal evolution times of t1max,1H = 10 ms and t2max,1H = 73.5 ms, spectral widths of SW1,1H = 22.2 ppm and SW2,1H = 22.4 ppm, an interscan delay of 1.3 s and 64 scans per increment resulting in a measurement time of ~ 10 h per spectrum. The unlabeled sample in D2O was 1.7 mM in 20 mM sodium phosphate, 0.4 mM EDTA, and was in ~ 100% D2O. The buildups for the sample in D2O were measured using a 2D PreSat [1H-1H] NOESY with four mixing times (40, 80, 120, and 160 ms). The spectra were acquired using a linear sampling scheme with 400(t1) × 1470(t2) complex points, maximal evolution times of t1max,1H = 20 ms and t2max,1H = 73.5 ms, spectral widths of SW1,1H = 22.2 ppm and SW2,1H = 22.4 ppm, an interscan delay of 1.5 s and 32 scans per increment resulting in a measurement time of ~ 10 h per spectrum.
Re-sampling of uniformly sampled data to NUS schedules and data processing for the 14mer RNA UUCG tetraloop
The uniformly sampled data from the UUCG tetraloop was resampled according to Poisson gap schedules (90% through 10%) and the data reconstructed in the same manner as for Pin1 and the WW domain except in 2D fashion with 200 and 400 complex points in the indirect dimensions for the H2O and D2O samples, respectively.
Simulation of Pin1 spectra with decreasing peak numbers
The simulated data was generated as follows: 3D peak detection was performed on a Pin1 protein uniformly sampled eNOE spectrum with a 24 ms mixing time and 8,000 total increments, and the diagonal signals were identified to provide a collection of 378 1H,13C 2D coordinates and peak heights. The 2D coordinates were used to make simulated 3D time-domain data where all of the signals occur at a single location in the directly-detected dimension (e.g., all the signals are in the same indirect plane). Simulations were generated by the simTimeND utility of NMRPipe, which generates time-domain data in NMRPipe format (Ying et al. 2017; Delaglio 1995). Time-domain data was simulated with the same digital resolution and spectral windows as the measured spectrum. Decays and unresolved couplings for the simulation were chosen to yield spectra that had a similar visual appearance to measured data. Time-domain exponential decays were set to vary randomly from 1H 10.5 Hz to 15.5 Hz, and 13C 16.2 Hz to 27.0 Hz, with random phase distortions in the range of ± 3° in both indirect dimensions. Each simulated peak had two unresolved couplings in each indirect dimension: the 1H indirect dimension had a fixed coupling of 4 Hz, and a random coupling in the range of 4 Hz to 14 Hz, and the 13C indirect dimension had a fixed coupling of 35 Hz, and a random coupling in the range of 7 Hz to 11 Hz. Gaussian random noise was added to the time domain such that the observed noise level in the corresponding fully sampled spectrum was about 0.25% of maximum intensity.
NOESY buildup fitting and distance restraints using CYANA
The NOESY spectra with the longest mixing time (56 ms for Pin1, 60 ms for the WW domain, and 160 ms for the UUCG tetraloop) were assigned in ccpNMR (Vranken 2005), the peak lists exported to NMRPipe format, and then cross- and diagonal-peak intensities at all mixing times were extracted using the NlinLS autofit script in NMRPipe for all NUS percentages. Fitted auto-relaxation rate constant (ρ) and initial magnetization (M0) values were used to determine cross-relaxation rate constants (σ) using the full-matrix approach (Orts et al. 2012) which is part of the eNORA2 (Strotz et al. 2017) package and has also been implemented into CYANA (Güntert et al. 1997; Güntert and Buchner 2015). Given the large number of data sets to be analyzed, we used CYANA because fitting the buildups is user friendly and extracting eNOE distances is completely automated. Spin-diffusion corrections were calculated from NMR structures of full-length Pin1 determined in our laboratory (to be published elsewhere), the WW domain (Strotz et al. 2020), as well as the UUCG tetraloop (Nichols 2018b) and applied to the intensities of the cross-peak buildup curves. The spin diffusion corrections and eNOE distances depend on the overall correlation time τc of the molecule via the spectral density function (Vögeli 2014a). A τc value of 4.25 ns, determined from R1/R1ρ measurements, was used for the WW domain (Strotz et al. 2015) and an average ρ value of 5.3 s−1 was used for initial fit determination. For the UUCG tetraloop, we used a τc value of 2.23 ns (Nichols 2018b) and average ρ value of 5.0 s−1. Because full-length Pin1 has two distinct domains with a flexible linker, the eNOE buildup fits had to be split into three separate calculations and then combined together. For each NUS percentage, the eNOE analysis was carried out with the relevant parameters for the WW domain, the linker, and the PPIase domain, and then the rates and related distances were combined to form master files. The τc values used were 14.1 ns for the PPIase domain, 11.3 ns for the WW domain, and 3.6 ns for the linker, which were determined from R1/R1ρ measurements carried out on the full-length Pin1 sample (to be published elsewhere), and the average ρ value was 10.18 s−1. For Pin1 and the UUCG tetraloop, all uni-directional buildups were normalized to the spin of origin (i → j) except when the diagonal peak of origin was missing or overlapped. In this case the uni-directional eNOE was normalized to the spin of destination (j → i), if it was present and of good quality (Strotz et al. 2015). Note that the optimal choice of the diagonal peaks used for normalization depends on the pulse sequence chosen, but generally the origin and destination peaks show a similar performance (Strotz et al. 2015). For the WW domain, this was reversed, meaning that uni-directional eNOEs were normalized to the spin of destination (j → i) and those peaks with a missing diagonal peak of destination were normalized to the spin of origin (i → j). The quality of the fits was inspected visually and an upper limit χN value was selected, above which the eNOE buildups and extracted distances were discarded automatically. This value was 29,000 for Pin1, 27,500 for the WW domain, and 35,000 for the UUCG tetraloop. In general, χN values higher than the selected cutoffs corresponded to peaks which no longer had sufficiently large signal-to-noise to be visible in the spectrum at that NUS density. Previously determined error tolerances for bi- and uni-directional eNOEs (Strotz et al. 2017) were automatically applied by CYANA. Generic normalized eNOEs (gn-eNOEs), which are loose upper limit restraints derived from cross peaks which cannot be normalized to a diagonal peak (Chi 2015a, b), were created by supplying upper limit M0 and average ρ values calculated from each of the relevant atom types (HA, HB1, HB* for proteins, and H1, H8, H1′ for RNA … etc.) and given an error tolerance of 20%.
Structure calculations in CYANA
For Pin1, the bi- and uni-directional eNOEs and gn-eNOEs from each of the domain-specific calculations (WW, linker, and PPIase) were sorted into combined upper and lower limit distance restraint files for all NUS percentages and used as input for structure calculations in CYANA 3.98 (Güntert et al. 1997; Güntert and Buchner 2015). The eNOE distance restraints were also supplemented with previously determined 3JHN-Hβ and 3JHα-Hβ scalar couplings (Born et al. 2018) as well as stereospecific assignments (Orts et al. 2013). For the UUCG tetraloop, bi- and uni-directional eNOEs and gn-eNOEs involving exchangeable protons derived from the H2O NOESY series were added to those derived from the D2O series and input into CYANA. For the WW domain, only bi- and uni-directional eNOEs were used as well as previously determined stereospecific assignments (Strotz et al. 2015). The calculations started with 100 initial structures with random torsion angle values using the standard simulated annealing protocol with 50,000 torsion angle dynamics steps. The 20 structures with the lowest target function values were selected for the ensembles. Distance restraints that violated the mean structures by more than 0.6 Å were discarded and the structures were re-calculated. Mean structures were created using Molmol (Koradi et al. 1996). Root-mean-square deviations (RMSDs) were calculated using CYANA. Backbone RMSD values were reported for Pin1 and the WW domain and heavy-atom RMSDs for the UUCG tetraloop. For Pin1, the terminal residues of the domains as well as a flexible loop in the WW domain were excluded from the RMSD calculation (the residues used were 4–15, 23–36 for the WW domain and 53–161 for the PPIase domain). For the WW domain and 14mer UUCG tetraloop residues 8–33 and nucleotides 2–13 were used to calculate RMSD values, respectively.
References
Aoto PC, Fenwick RB, Kroon GJA, Wright PE (2014) Accurate scoring of non-uniform sampling schemes for quantitative NMR. J Magn Reson. https://doi.org/10.1016/j.jmr.2014.06.020
Barna JC, Laue E, Mayger M, Skilling J, Worrall SJ (1987) Exponential sampling, an alternative method for sampling in two-dimensional NMR experiments. J Magn Reson. https://doi.org/10.1016/0022-2364(87)90225-3
Boelens R, Koning TMG, Kaptein R (1988) Determination of biomolecular structures from proton-proton NOE’s using a relaxation matrix approach. J Mol Struct. https://doi.org/10.1016/0022-2860(88)80062-0
Boelens R, Koning TMG, van der Marel GA, van Boom JH, Kaptein R (1989) Iterative procedure for structure determination from proton-proton NOEs using a full relaxation matrix approach. Application to a DNA octamer. J Magn Reson 82:290–308
Born A et al (2018a) Backbone and side-chain chemical shift assignments of full-length, apo, human Pin1, a phosphoprotein regulator with interdomain allostery. Biomol NMR Assign 13:85
Born A et al (2018b) Efficient stereospecific Hβ2/3 NMR assignment strategy for mid-size proteins. Magnetochemistry 4:25
Bostock MJ, Holland DJ, Nietlispach D (2012) Compressed sensing reconstruction of undersampled 3D NOESY spectra: application to large membrane proteins. J Biomol NMR. https://doi.org/10.1007/s10858-012-9643-4
Brüschweiler R et al (1992) Influence of rapid intramolecular motion on NMR cross-relaxation rates. A molecular dynamics study of antamanide in solution. J Am Chem Soc. https://doi.org/10.1021/ja00033a002
Bürgi R, Pitera J, van Gunsteren WF (2001) Assessing the effect of conformational averaging on the measured values of observables. J Biomol NMR. https://doi.org/10.1023/A:1011295422203
Chi CN et al (2015a) A structural ensemble for the enzyme cyclophilin reveals an orchestrated mode of action at atomic resolution. Angew Chemie Int Ed 54:11657–11661
Chi CN, Strotz D, Riek R, Vögeli B (2015b) Extending the eNOE data set of large proteins by evaluation of NOEs with unresolved diagonals. J Biomol NMR 62:63–69
Chi CN, Strotz D, Riek R, Vögeli B (2018) NOE-derived methyl distances from a 360 kDa proteasome complex. Chem A Eur J 24:2270–2276
Delaglio F et al (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Güntert P, Buchner L (2015) Combined automated NOE assignment and structure calculation with CYANA. J Biomol NMR 62:453–471
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program Dyana. J Mol Biol 273:283–298
Hansen DF (2019) Using deep neural networks to reconstruct non-uniformly sampled NMR spectra. J Biomol NMR. https://doi.org/10.1007/s10858-019-00265-1
Hiller S, Ibraghimov I, Wagner G, Orekhov VY (2009) Coupled decomposition of four-dimensional NOESY spectra. J Am Chem Soc. https://doi.org/10.1021/ja902012x
Hoch JC (1989) Modern spectrum analysis in nuclear magnetic resonance: alternatives to the Fourier transform. Methods Enzymol. https://doi.org/10.1016/0076-6879(89)76014-6
Holland DJ, Bostock MJ, Gladden LF, Nietlispach D (2011) Fast multidimensional NMR spectroscopy using compressed sensing. Angew Chemie Int Ed. https://doi.org/10.1002/anie.201100440
Hyberts SG, Frueh DP, Arthanari H, Wagner G (2009) FM reconstruction of non-uniformly sampled protein NMR data at higher dimensions and optimization by distillation. J Biomol NMR. https://doi.org/10.1007/s10858-009-9368-1
Hyberts SG, Takeuchi K, Wagner G (2010) Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J Am Chem Soc. https://doi.org/10.1021/ja908004w
Hyberts SG, Milbradt AG, Wagner AB, Arthanari H, Wagner G (2012a) Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J Biomol NMR. https://doi.org/10.1007/s10858-012-9611-z
Hyberts SG, Arthanari H, Wagner G (2012b) Applications of non-uniform sampling and processing. Top Curr Chem. https://doi.org/10.1007/128_2011_187
Hyberts SG, Robson SA, Wagner G (2013) Exploring signal-to-noise ratio and sensitivity in non-uniformly sampled multi-dimensional NMR spectra. J Biomol NMR. https://doi.org/10.1007/s10858-012-9698-2
Hyberts SG, Arthanari H, Robson SA, Wagner G (2014) Perspectives in magnetic resonance: NMR in the post-FFT era. J Magn Reson. https://doi.org/10.1016/j.jmr.2013.11.014
Hyberts SG, Robson SA, Wagner G (2017) Interpolating and extrapolating with hmsIST: seeking a t max for optimal sensitivity, resolution and frequency accuracy. J Biomol NMR 68:139–154
Jaravine VA, Zhuravleva AV, Permi P, Ibraghimov I, Orekhov VY (2008) Hyperdimensional NMR spectroscopy with nonlinear sampling. J Am Chem Soc. https://doi.org/10.1021/ja077282o
Kalk A, Berendsen HJC (1976) Proton magnetic relaxation and spin diffusion in proteins. J Magn Reson. https://doi.org/10.1016/0022-2364(76)90115-3
Keepers JW, James TL (1984) A theoretical study of distance determinations from NMR. Two-dimensional nuclear overhauser effect spectra. J Magn Reson 57:404–426
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14:51–55
Kumar A, Wagner G, Ernst RR, Wuethrich K (1981) Buildup rates of the nuclear Overhauser effect measured by two-dimensional proton magnetic resonance spectroscopy: implications for studies of protein conformation. J Am Chem Soc 103:3654–3658
Lu KP, Hanes SD, Hunter T (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. https://doi.org/10.1038/380544a0
Monajemi H (2016) Phase transitions in deterministic compressed sensing, with applications to magnetic resonance spectroscopy. Stanford University. https://doi.org/10.13140/RG.2.2.21686.80960
Monajemi H, Donoho DL (2018) Sparsity/undersampling tradeoffs in anisotropic undersampling, with applications in MR imaging/spectroscopy. Inf Inference A J IMA 00:1–46
Neuhaus D, Williamson M (2000) The nuclear overhauser effect in structural and conformational analysis. New York: Cambridge
Nichols P et al (2017) The exact nuclear overhauser enhancement: recent advances. Molecules 22:1176
Nichols PJ et al (2018a) Extending the applicability of exact nuclear overhauser enhancements to large proteins and RNA. ChemBioChem. https://doi.org/10.1002/cbic.201800237
Nichols PJ et al (2018b) High-resolution small RNA structures from exact nuclear overhauser enhancement measurements without additional restraints. Nat Commun Biol. https://doi.org/10.1038/s42003-018-0067-x
Nozinovic S, Fürtig B, Jonker HRA, Richter C, Schwalbe H (2009) High-resolution NMR structure of an RNA model system: the 14-mer cUUCGg tetraloop hairpin RNA. Nucleic Acids Res 38:683–694
Orekhov VY, Ibraghimov I, Billeter M (2003) Optimizing resolution in multidimensional NMR by three-way decomposition. J Biomol NMR. https://doi.org/10.1023/A:1024944720653
Orts J, Vögeli B, Riek R (2012) Relaxation matrix analysis of spin diffusion for the NMR structure calculation with eNOEs. J Chem Theory Comput 8:3483–3492
Orts J, Vögeli B, Riek R, Güntert P (2013) Stereospecific assignments in proteins using exact NOEs. J Biomol NMR. https://doi.org/10.1007/s10858-013-9780-4
Palmer MR et al (2015) Sensitivity of nonuniform sampling NMR. J Phys Chem B. https://doi.org/10.1021/jp5126415
Post CB (1992) Internal motional averaging and three-dimensional structure determination by nuclear magnetic resonance. J Mol Biol. https://doi.org/10.1016/0022-2836(92)90471-U
Schmieder P, Stern AS, Wagner G, Hoch JC (1994) Improved resolution in triple-resonance spectra by nonlinear sampling in the constant-time domain. J Biomol NMR. https://doi.org/10.1007/BF00156615
Solomon I (1955) Relaxation processes in a system of two spins. Phys Rev 99:559–565
Stern AS, Hoch JC (2015) A new approach to compressed sensing for NMR. Magn Reson Chem 53:908–912
Stern AS, Li KB, Hoch JC (2002) Modern spectrum analysis in multidimensional NMR spectroscopy: comparison of linear-prediction extrapolation and maximum-entropy reconstruction. J Am Chem Soc. https://doi.org/10.1021/ja011669o
Stern AS, Donoho DL, Hoch JC (2007) NMR data processing using iterative thresholding and minimum l1-norm reconstruction. J Magn Reson 188:295–300
Strotz D, Orts J, Minges M, Vögeli B (2015) The experimental accuracy of the uni-directional exact NOE. J Magn Reson 259:32–46
Strotz D, Orts J, Chi CN, Riek R, Vögeli B (2017) ENORA2 exact NOE analysis program. J Chem Theory Comput 13:4336–4346
Strotz D, Orts J, Kadavath H, Friedmann M, Ghosh D, Olsson S, Chi C, Pokharna A, Güntert P, Vögeli B, Riek R (2020) Protein allostery at atomic resolution. Angew Chem Int Engl Ed. https://doi.org/10.1002/anie.202008734
Vögeli B (2014a) The nuclear Overhauser effect from a quantitative perspective. Prog Nucl Magn Reson Spectrosc 78:1–46
Vögeli B et al (2009) Exact distances and internal dynamics of perdeuterated ubiquitin from NOE buildups. J Am Chem Soc 131:17215–17225
Vögeli B, Friedmann M, Leitz D, Sobol A, Riek R (2010) Quantitative determination of NOE rates in perdeuterated and protonated proteins: practical and theoretical aspects. J Magn Reson 204:290–302
Vögeli B, Güntert P, Riek R (2013) Multiple-state ensemble structure determination from eNOE spectroscopy. Mol Phys 111:437–454
Vögeli B et al (2014b) Towards a true protein movie: a perspective on the potential impact of the ensemble-based structure determination using exact NOEs dedicated to Prof Dr Christian Griesinger. J Magn Reson 241:53–59
Vögeli B, Olsson S, Güntert P, Riek R (2016) The Exact NOE as an alternative in ensemble structure determination. Biophys J 110:113–126
von Schlippenbach T, Oefner PJ, Gronwald W (2018) Systematic evaluation of non-uniform sampling parameters in the targeted analysis of urine metabolites by 1H,1H 2D NMR spectroscopy. Sci Rep. https://doi.org/10.1038/s41598-018-22541-0
Vranken WF et al (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct Funct Genet 59:687–696
Wüthrich, K. NMR of Proteins and Nucleic Acids. 32, (Wiley, 1986).
Ying J, Delaglio F, Torchia DA, Bax A (2017) Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J Biomol NMR. https://doi.org/10.1007/s10858-016-0072-7
Zinovjev K, Liepinsh E (2013) Validation of the CHARMM27 force field for nucleic acids using 2D nuclear overhauser effect spectroscopy. J Biophys Chem 04:58–65
Acknowledgements
This research is funded by NSF Grant 1917254 for Infrastructure Innovation for Biological Research and a start-up package from the University of Colorado to B.V.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nichols, P.J., Born, A., Henen, M.A. et al. Reducing the measurement time of exact NOEs by non-uniform sampling. J Biomol NMR 74, 717–739 (2020). https://doi.org/10.1007/s10858-020-00344-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-020-00344-8