
Abstract
The measurement of long-range distances remains a challenge in solid-state NMR structure determination of biological macromolecules. In 2D and 3D correlation spectra of uniformly 13C-labeled biomolecules, inter-residue, inter-segmental, and intermolecular 13C–13C cross peaks that provide important long-range distance constraints for three-dimensional structures often overlap with short-range cross peaks that only reflect the covalent structure of the molecule. It is therefore desirable to develop new approaches to obtain spectra containing only long-range cross peaks. Here we show that a relaxation-compensated modification of the commonly used 2D 1H-driven spin diffusion (PDSD) experiment allows the clean detection of such long-range cross peaks. By adding a z-filter to keep the total z-period of the experiment constant, we compensate for 13C T1 relaxation. As a result, the difference spectrum between a long- and a scaled short-mixing time spectrum show only long-range correlation signals. We show that one- and two-bond cross peaks equalize within a few tens of milliseconds. Within ~200 ms, the intensity equilibrates within an amino acid residue and a monosaccharide to a value that reflects the number of spins in the local network. With T1 relaxation compensation, at longer mixing times, inter-residue and inter-segmental cross peaks increase in intensity whereas intra-segmental cross-peak intensities remain unchanged relative to each other and can all be subtracted out. Without relaxation compensation, the difference 2D spectra exhibit both negative and positive intensities due to heterogeneous T1 relaxation in most biomolecules, which can cause peak cancellation. We demonstrate this relaxation-compensated difference PDSD approach on amino acids, monosaccharides, a crystalline model peptide, a membrane-bound peptide and a plant cell wall sample. The resulting difference spectra yield clean multi-bond, inter-residue and intermolecular correlation peaks, which are often difficult to resolve in the parent 2D spectra.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the most important elements in biomolecular structure determination using solid-state NMR spectroscopy is the measurement of long-range distances, which define the three-dimensional fold of proteins, sidechain packing, relative orientations between two domains in a macromolecule, and intermolecular interfaces (Hong 2006; Hong et al. 2012). To date, the measurement of such long-range distances, operationally defined as distances longer than about 5 Å, has been mostly conducted using through-space 13C correlation experiments, which give cross peaks whose intensities qualitatively reflect internuclear distances. The most commonly used correlation experiment is the 1H-driven spin diffusion (PDSD) experiment, which can be conducted in 2D or 3D, with one or two mixing times (Li et al. 2010; Meier 1994; Takegoshi et al. 2001). In these spectra, both one-bond and two-bond correlations, which are useful for resonance assignment but not distance extraction, and long-range correlations with high structural content, are present. The former usually have much higher intensities than the latter, thus making it difficult to assign and extract the structurally informative long-range distances (Miao et al. 2013).
This problem is partly alleviated by the PAR experiment developed by Griffin and coworkers (De Paëpe et al. 2008; Lewandowski et al. 2009), which increases the intensities of long-range cross peaks relative to the short-range ones by attenuating the dipolar truncation mechanism through trilinear coherence terms between 13C and 1H spins. It is also reduced by selective isotopic labeling (Castellani et al. 2002; Higman et al. 2009; Hong 1999; Hong and Jakes 1999; Loquet et al. 2011), which reduces the number of directly bonded 13C–13C spin pairs in the protein. However, no experiment so far exists to completely remove the short-range cross peaks in the spectra of uniformly 13C-labeled biomolecules.
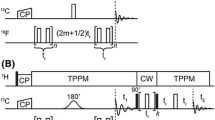
In this work, we introduce a simple T1-relaxation-compensated difference PDSD experiment that allows the clean detection of long-range cross peaks without overlap from short-range cross peaks for 13C-labeled proteins, polysaccharides and other biomolecules. The term “short range” here includes two scenarios: one- and two-bond cross peaks on the one hand, and intra-residue, mostly multi-bond, cross peaks on the other. The one- and two-bond cross peaks can be suppressed by subtracting a spectrum measured with mixing times on the order of ~30 ms from spectra measured with mixing times on the order of a few hundred milliseconds, while suppression of all intra-residue cross peaks requires, as we show below, subtraction of a spectrum measured with intermediate mixing times of ~100 ms from spectra measured with very long mixing times of ~1 s. Since the shortest 13C T1 relaxation times of methyl-containing biomolecules are on the order of a few hundred milliseconds, the difference spectra obtained from the second scenario are subject to significant and differential T1 relaxation effects, giving positive difference peaks for the slow-relaxing species, which are desirable, and negative difference peaks for the fast-relaxing moieties, which are undesirable because they reflect mostly short-range correlations. We address this problem by adding a z-filter before the 13C chemical shift evolution period to make the total z-period constant (Fig. 1). This relaxation compensation principle was first introduced in the CODEX technique (deAzevedo et al. 2000; Schmidt-Rohr et al. 2002); the current implementation differs by not involving any recoupling of anisotropic spin interactions.

Pulse sequence for the relaxation-compensated PDSD experiment. The sum of the z-filter and the mixing time is kept constant while the mixing times are varied
In addition, we consider what pairs of mixing times will give only the desired long-range cross peaks in the difference spectrum. At very short mixing times of ~10 ms, two-bond cross peaks still rise in intensity while one-bond signals are already past their maximum. These factors cause both positive and negative cross peaks in the difference spectrum (Miao et al. 2013), which can cause partial peak cancellation in a congested spectrum. By choosing both mixing times to be longer (≥30 ms) than the one- and two-bond transfer rates, we ensure that the intensities of the one- and two-bond cross peaks remain unchanged relative to each other but their total intensities decrease together at the longer mixing time due to magnetization loss to longer-range correlations. Thus in a suitably scaled difference spectrum, all short-range cross-peaks will be removed.
We demonstrate this relaxation-compensated difference PDSD method on several small-molecule model compounds, a membrane peptide and plant cell walls, and show that the difference spectra give uniformly positive long-range correlation signals that are multi-bond, inter-residue, or intermolecular. For comparison, difference spectra obtained without relaxation compensation exhibit mixed negative and positive cross peaks. To identify the best mixing times for difference spectroscopy, we measured the spin diffusion buildup curves of the model compounds, which are seen to equilibrate to a value that reflects the number of spins in the local network, thus providing useful structural information.
Materials and methods
Four uniformly 13C-labeled model compounds, histidine (His), glutamine (Gln), the tripeptide formyl-Met-Leu-Phe-OH (MLF), and d-glucose, were used to examine the spin diffusion buildup of the relaxation-compensated PDSD experiment. The pH 6.0 histidine sample contains both cationic histidine and the neutral τ tautomer (Li and Hong 2011). The tripeptide MLF (Rienstra et al. 2000, 2002) has 20 13C labels and allows us to assess the performance of the difference PDSD experiment in a large spin network analogous to uniformly 13C, 15N-labeled proteins. A site-specifically labeled influenza M2 transmembrane peptide (M2TM) sample was used to investigate the determination of long-range distances in membrane proteins. The peptide contains 13C, 15N-labeled residues at G34, H37, and I42 (Hu et al. 2010), and is bound to a virus-mimetic lipid membrane (Luo et al. 2009) at pH 4.5, which corresponds to the open state of this proton channel. We also applied the relaxation-compensated PDSD experiment to uniformly 13C-labeled Arabidopsis thaliana primary cell wall (Dick-Perez et al. 2011; Wang et al. 2012), which contains a mixture of polysaccharides and glycoproteins. The plant was 13C-labeled by growth in the dark in liquid culture containing 13C-labeled glucose as the only carbon source. The seedlings were harvested, and soluble molecules, lipids, intracellular proteins, and starch were removed by treatments with chloroform/ethanol (1:1) solution, pH 5.2 sodium acetate buffer containing SDS and sodium metabisulfite, and α-amylase, respectively. The remaining alcohol-insoluble material is the cell wall.
Solid-state NMR experiments
Histidine, glutamine, MLF, and M2TM spectra were measured on a Bruker 400 MHz spectrometer, while glucose and plant cell wall spectra were measured on a Bruker 600 MHz NMR spectrometer. Typical radiofrequency field strengths were ~70 kHz for 1H decoupling and 50 kHz for 13C pulses. All 13C chemical shifts were externally referenced to the adamantane CH2 peak at 38.48 ppm on the TMS scale.
The pulse sequence for the relaxation-compensated PDSD experiment (Fig. 1) contains a z-filter before the evolution period, and the sum of the z-filter (tz) and mixing time (tm) is kept constant to compensate for T1 relaxation. Glutamine, histidine and MLF spectra were measured at room temperature using constant z-periods of 0.505, 1.005, and 1.505 s, respectively, and the MAS frequencies ranged from 7 to 10 kHz (Table S1). The M2TM spectra were measured with a constant z-period of 1.505 s at 273 K under 9 kHz MAS. The glucose and plant cell wall samples were measured using a constant z-period of 1.005 s under 8 kHz MAS. The temperature was 273 K for glucose and 253 K for the plant cell wall.
The difference spectra were obtained by subtracting a short mixing-time spectrum from a long mixing-time spectrum, with an adjustable scaling factor for the former. For the histidine difference spectrum, no scaling was applied. For glucose, the difference spectrum between 1.0 and 0.2 s involved scaling the latter by 0.95 to give null intensities, while the difference spectrum between 200 and 20 ms involved scaling the 20 ms spectrum by 0.78 to remove the one-bond cross peaks. For MLF, a difference spectrum between 300 and 30 ms used a scaling factor of 0.35 to remove one-bond cross peaks. For influenza M2TM, a scaling factor of 0.70 was applied to the 100 ms spectrum before subtraction from the 1.5 s spectrum. For the plant cell wall sample, the relaxation-compensated difference between the 1.0 and 0.2 s spectra used a scaling factor of 0.78 for the latter, while a regular PDSD difference spectrum used a scaling factor of 0.69 for the 0.2 s spectrum.
Results and discussion
Small-molecule model compounds
We first demonstrate the T1-compensated PDSD experiment on model compounds containing a small number of 13C spins. We monitor the spin diffusion buildup behavior of cross peaks to identify the magnetization equilibration times. To make the cross-peak intensity values meaningful, we divide the integrated area of a peak by the sum of all peak areas in the same ω1 cross section:
When spin diffusion in a local network of n spins is complete, the cross-peak intensity should equilibrate to 1/n. This normalization procedure is advantageous over a previous method that involves double normalization by comparison with a short-mixing spectrum (Wang et al. 2012, 2013). As shown for glutamine (Fig. S1), Eq. (1) yields an equilibrium intensity of 0.20 for all carbons in this five-carbon molecule, while the double-normalization method gives rise to an arbitrary equilibrium value of 0.43.
Figure 2 shows the relaxation-compensated PDSD buildup curves of histidine. Using a total z-period of 1.005 s and mixing times from 5 ms to 1.0 s, we obtained an equilibrium intensity of ~ 17 % for all Cα cross peaks, consistent with the presence of six carbons in this molecule. The exponential time constants for the initial buildup range from 2 ms for one-bond cross peaks to about 50 ms for the longest-range cross peak between backbone Cα and imidazole Cε1. The time constants increase with the number of intervening bonds and the results are similar between cationic and neutral histidine, as expected. Importantly, once equilibrated, the 13C magnetization does not decay further with mixing time, confirming that T1 relaxation effects have been compensated for. The one-bond Cα–CO cross peak has a slower initial buildup time constant (4.8 ms) than the one-bond Cα–Cβ peak (2 ms), but the two peaks have the same decay constants (Fig. S2). As a result, the Cα–CO intensity overshoots while the Cα–Cβ intensity does not. We attribute the slower buildup of the Cα–CO cross peak to the lack of a proton on CO, which slows down 1H-driven 13C spin diffusion compared to the protonated Cβ.

Relaxation-compensated PDSD buildup of histidine, measured with a total z-period of 1.005 s. Only the cationic histidine data is shown, but neutral histidine shows the same behavior. a Buildup time constants from Cα to other carbons. b ω2 cross sections of cationic histidine Cα at 54 ppm, scaled to the Cα peak intensity. c Spin diffusion buildup curves. The peaks equilibrate to a value of 1/6, consistent with the presence of six carbons in the molecule. The buildup time constants increase with the number of bonds separating the two carbons
Based on these buildup curves, we obtained a histidine difference 2D spectrum by subtracting the 100 ms spectrum from the 1.0 s spectrum. Since intermolecular contacts between cationic and neutral histidines are much longer than the molecular length scale, no cross peaks are expected between the different molecules and all intramolecular cross peaks should be null in the difference spectrum. This is confirmed in Fig. 3, which shows suppression of most peaks except for weak residual intensities for the Cε1–Cα and Cε1–CO peaks, which have not fully equilibrated by 100 ms due to the long distances between these carbons. No scaling factor was applied to the 100 ms spectrum, verifying that relaxation compensation and the choice of the two mixing times longer than the magnetization equilibration time permit clean difference spectroscopy.

Relaxation-compensated 2D PDSD spectra of histidine, measured with a total z-period of 1.005 s. a 1 s mixing. b 100 ms mixing. c Difference spectrum, obtained without scaling the 100 ms spectrum. Negative intensities are plotted in green. Most intra-residue cross peaks are suppressed, except for weak Cε1–Cα and Cε1–CO peaks in the 136-ppm cross section due to the long-range nature of these cross peaks
Because of its central role in carbohydrate chemistry, glucose is examined as a model compound for the relaxation-compensated PDSD experiment. In the 6-carbon pyranose ring, the magnetization is expected to equilibrate to 1/6. Indeed, all resolved 13C signals reach an equilibrium intensity of 17 % (Fig. 4), while the overlapped C3/C4 peak gave twofold higher equilibrium intensity (32 %). The buildup constants range from 2.4 ms for one-bond correlations to ~30 ms for the C1–C6 correlation. Based on these buildup curves, we obtained a difference spectrum between 1.0 s and 200 ms, using a scaling factor of 0.95 for the latter. The difference spectrum is clean, showing no cross peaks (Fig. 5a), indicating that all intramolecular contacts have equilibrated by 200 ms and T1 relaxation effects are corrected for. To remove one-bond cross peaks and detect only multi-bond cross peaks, we obtained a difference spectrum between the 200 and 20 ms spectra using a scaling factor of 0.78 for the latter (Fig. 5b, c). After 20 ms, the C1–C2 cross peak intensity still decays due to magnetization transfer to other carbons (Fig. 4c). Thus, the 20 ms C1–C2 intensity is higher than the equilibrium intensity, which accounts for the scaling factor.

Relaxation-compensated PDSD data and buildup curves of d-glucose, measured with a total z-period of 1.005 s. a Buildup time constants of α-d-glucose. b The C1–C1 diagonal peak equilibrates to 17 %. c Buildup curves of C1–Cx cross peaks, with equilibrium intensities of 17 % for the resolved peaks and 32 % for the doubly overlapped C1–C3, 4 peak. d Buildup time constants of β-d-glucose

Relaxation-compensated 2D PDSD spectra of d-glucose. a 1.0 s spectrum, 200 ms spectrum, and difference spectrum, obtained with a scaling factor of 0.95 for the 200 ms spectrum. No cross peaks are observed in the difference spectrum, corresponding to magnetization equilibration by 200 ms. b 200 ms spectrum, 20 ms spectrum, and difference spectrum, obtained with a scaling factor of 0.78 for the 20 ms spectrum. All one-bond cross peaks are removed while multi-bond cross peaks remain. c Representative cross sections of the 200, 20 ms and difference spectra. One-bond cross peaks (red dashed lines) have null difference intensities. The overlapped 1-2/3β peak shows residual difference intensity due to the two-bond 1-3β correlation
Uniformly 13C-labeled polysaccharides and site-specifically labeled membrane peptides
Since 13C magnetization within a monosaccharide equilibrates by 200 ms (Fig. 4), longer-range correlations will manifest specifically at times longer than 200 ms. Fig. S3a, b show the 2D PDSD spectra of Arabidopsis primary cell walls, measured with a total z-period of 1.005 s. The difference spectrum between 1.0 and 0.2 s, with a scaling factor of 0.78 for the latter, shows the suppression of most protein cross peaks, and only long-range correlation peaks remain in the difference spectrum (Fig. 6a). For example, the (89, 84 ppm) cross peak results from the spatial contacts between interior cellulose C4 (iC4) and surface cellulose (sC4) or xyloglucan backbone C4 (GC4). The scaling factor results from the fact that the iC4–iC6 cross peak decays between 200 and 500 ms due to magnetization loss to other carbons.

Difference 2D PDSD spectra of the Arabidopsis primary cell wall, obtained by subtracting the 200 ms spectrum from the 1.0 s spectrum. a Relaxation-compensated difference spectrum, obtained using a scaling factor of 0.78 for the 200 ms spectrum. b Regular PDSD difference spectrum, obtained using a scaling factor of 0.69 for the 200 ms spectrum. The relaxation-compensated difference spectrum shows only intermolecular correlations, while the regular PDSD difference spectrum has negative intramolecular protein and pectin intensities due to their fast T1 relaxation rates. Both difference spectra are plotted using contour parameters of lev0 = 4, toplev = 100 and nlev = 16 in the Topspin software. c Representative 1D cross sections of cellulose, matrix polysaccharides and proteins. The relaxation-compensated difference spectra (top row) show positive intermolecular cross peaks between cellulose and pectins, which are difficult to resolve in the parent spectra. The regular PDSD difference spectra (bottom row) show a mixture of negative and positive cross peaks due to heterogeneous T1 relaxation. Assignments are indicated in red for cellulose, blue for matrix polysaccharides, and black for overlapped peaks
T1 relaxation compensation ensures that the difference PDSD spectrum shows only positive cross peaks. The only negative intensities appear along the diagonal, because magnetization continues to diffuse to other carbons with increasing mixing time. Having uniformly positive cross peaks is important, since in a crowded spectrum, the presence of both positive and negative intensities will cause partial peak cancellation. Without relaxation compensation, spectral subtraction fails, because functional groups with different T1 relaxation times will exhibit different extents of intensity decay at long mixing times. Figure 6b shows the difference spectrum obtained from regular PDSD experiments. Strong negative intensities for protein and matrix polysaccharide peaks are observed due to much shorter 13C T1 relaxation times of these molecules compared to cellulose. This heterogeneous T1 relaxation means that no single scaling factor allows the satisfactory removal of all short-range cross peaks. For the difference spectrum in Fig. 6b, we used a scaling factor that removed the cellulose intramolecular cross peaks (Fig. S3d–f), which gave negative intensities for the fast-relaxing pectin and protein signals. Smaller scaling factors that remove the protein cross peaks will retain cellulose cross peaks in the difference spectrum. Therefore, relaxation compensation is important for detecting long-range correlations in dynamically heterogeneous samples.
The relaxation-compensated difference PDSD spectrum of the plant cell wall reveals intermolecular correlations between cellulose and pectic sugars such as galacturonic acid (GA) and rhamnose (R) (Fig. 6c), consistent with previously published 2D and 3D 13C–13C correlation spectra (Dick-Perez et al. 2011; Wang et al. 2012). These cross peaks indicate that pectins contact cellulose microfibrils within ~1 nm, the distance scale probed by 13C spin diffusion. The exact distances between cellulose and matrix polysaccharides are likely distributed, and this distribution is not known without high-resolution structures of the cell wall; however, we can obtain semi-quantitative information of the distances by examining the crystal structure of the Iβ crystalline allomorph of cellulose (Atalla and VanderHart 1984; Nishiyama et al. 2002), which is the dominant form of cellulose in plant cell walls. Minimal distance separations of ~5 Å are present between surface and interior glucan chains (Fig. S4a) (Wang et al. 2013), and the full distance distribution has significant intensities below 8 Å. Since the cellulose-matrix polysaccharide cross peaks have weaker intensities than the surface-interior cellulose cross peaks (Fig. 6c), the minimum distance separation between the cellulose surface and pectins is expected to be longer than ~5 Å.
In non-mixture samples, significant T1 relaxation differences can exist, especially when 13C labeling is not uniform and hence spin diffusion does not equilibrate the relaxation rates. For example, T1 relaxation is expected to be much faster for methyl-rich protein residues such as Ile and Leu than for aromatic residues. To examine the effectiveness of relaxation-compensated difference PDSD method for site-specifically labeled samples, we measured the PDSD spectra of GHI-M2TM. A difference spectrum was obtained between 1.5 and 100 ms mixing-time spectra, using a scaling factor of 0.70 for the latter. Figure 7 shows that all intra-residue cross peaks, including both the fast-relaxing methyl-rich I42 and the slow-relaxing aromatic moiety of H37 and the G34 backbone, are removed in the difference spectrum, while clear inter-residue cross peaks between G34 and H37 are retained. The resolution enhancement due to the difference spectroscopy is exemplified by the H37 Cα–G34 CO cross peak, which overlaps with the H37 Cα–CO cross peak in the parent spectra. High-resolution structures of the tetrameric M2TM helical bundle at both low and high pH (Acharya et al. 2010; Cady et al. 2010) indicate that the shortest G34–H37 distances are intrahelical and are between 4 and 5 Å (Fig. S4b), while interhelical G34–H37 distances are longer than 7 Å at low pH and longer than 5 Å at high pH. Thus, the observed G34–H37 cross peaks should mainly result from intrahelical contacts.

Relaxation-compensated 2D PDSD spectra of membrane-bound GHI-M2TM, using a total z period of 1.505 s. a 1.5 s mixing. b 100 ms mixing. The main intra-residue cross peaks are assigned. c Difference spectrum, obtained with a scaling factor of 0.70 for the 100 ms spectrum. Multiple long-range G34–H37 cross peaks are observed while intra-residue cross peaks are removed in the difference spectrum
Uniformly 13C-labeled peptides and proteins
To assess how well the relaxation-compensated difference PDSD method works for uniformly 13C labeled peptides and proteins, we measured the spectra of MLF, which contains 20 13C spins. Figure 8 shows that by ~500 ms, the 13C magnetization has equilibrated between Met and Phe, which have the longest-distance contacts in the peptide. Intra-residue spin diffusion has time constants of 3–89 ms, comparable to those in histidine. The time constants are distinct between one-bond and multi-bond correlations. Thus, for removing one-bond cross peaks, a difference spectrum can be readily obtained by subtracting, for example, the 30 ms spectrum from a longer-mixing-time spectrum. Figure 9c shows this difference spectrum, using a scaling factor of 0.35 for the 30 ms spectrum. A few diagonal peaks remain with weak negative intensities, but all cross peaks show positive intensities and are mostly inter-residue. For example, the difference spectrum reveals long-range cross peaks from Fβ to Mα, Mβ, and Mγ (in rows and columns at 37 ppm), which are not apparent in the regular PDSD spectra due to significant overlap between the Fβ and Mβ signals. This highlights the usefulness of our T1-compensated difference PDSD approach.

Relaxation-compensated PDSD buildup curves of MLF, obtained with a total z-period of 1.505 s. Only the Met Cα buildup curves are shown for clarity. a Overlay of the buildup curves of Mα–Mβ, Mα–Lβ, and Mα–Fβ for mixing times up to 1.5 s. b Initial trajectories up to 500 ms of individual buildup curves. Dashed lines in several panels in the top row reproduce the diagonal peak’s decay curve to show that the one- and two-bond decays track the diagonal peak decay. c Buildup time constants from Met Cα to other carbons and the corresponding internuclear distances. d A schematic of the magnetization transfer time course for a linear chain of spins illustrates how the cross-peak decays resemble the diagonal-peak decay increasingly closely for spins closer to the source. After a certain mixing time, the intensities of all spins within a certain distance decay similarly together and can be removed in a difference spectrum. The scaling factor in the subtraction results from the common decay of the diagonal, one- and two-bond peaks by that mixing time

Relaxation-compensated 2D PDSD spectra of MLF measured with a total z-period of 1.505 s. a 300 ms mixing. b 30 ms mixing. c Difference spectrum, obtained with a scaling factor of 0.35 for the 30 ms spectrum. The cross peaks in the difference spectrum are predominantly inter-residual. Blue dashed lines give the positions of the Met intra-residue cross peaks, and intensities at most of these intersections have been suppressed in the difference spectrum, confirming clean subtraction due to relaxation compensation and appropriate choice of the mixing times and the scaling factor. Projections of the 2D spectra are shown at the top and on the right to facilitate cross-peak identification. All spectra are plotted using contour parameters of lev0 = 4, toplev = 100 and nlev = 16 in the Topspin software
The buildup curves in Fig. 8 indicate that, to detect long-range cross peaks between a segment of three residues and another domain in a protein, the mixing times needed to remove all cross peaks within the tripeptide unit is ~500 ms. For this 20-carbon peptide, each 13C ideally should reach an equilibrium intensity of 5 %. The measured equilibrium intensities are quantitatively consistent with this prediction for all resolved signals. Where n peaks overlap, the final value is n times 5 %. For example, the doubly overlapped Mα–Lγ/δ cross peak equilibrates to ~10 % (Fig. 8b). The 5-carbon overlapped Mα–Fδ, ε, ζ cross peak equilibrates to 13.7 %, which is lower than the theoretical value of 25 %. We attribute this observation to the complex motion of the phenyl ring (Rienstra et al. 2002) and the large chemical shift anisotropies of aromatic carbons, which may slow down 13C spin diffusion.
Choice of mixing times and the scaling factor for difference spectroscopy
The above examples show that the relaxation-compensated PDSD experiment works well for site-specifically labeled biomolecules and uniformly labeled biomolecular mixtures, where the timescale of intra-segmental spin diffusion of ~100 ms is well separated from the inter-segmental or intermolecular spin diffusion timescale of ~1 s. This separation of timescale allows unambiguous detection of inter-residue and intermolecular correlations. In uniformly 13C-labeled non-mixture samples, complete magnetization equilibrium in the whole protein is irrelevant, instead one- and two-bond cross peaks should be removed to detect multi-bond and inter-residue cross peaks. Since 13C spin diffusion is more rapid in uniformly labeled molecules than in site-specifically labeled samples, the most suitable mixing times for difference spectroscopy should be a few tens of milliseconds and a few hundreds of milliseconds.
The difference spectra in Figs. 3c, 5b, 6a, 7c, and 9c show that removal of one-bond cross peaks always results in small, slightly negative residual diagonal peaks. This observation can be understood by a simple analysis of the spin-diffusion dynamics and is useful for determining the scaling factor without requiring a priori knowledge of which cross peaks are from one-bond couplings: if the diagonal in the difference spectrum is slightly negative (about −2 % of the full diagonal intensity), then the residual one-bond cross peaks will be very small.
We consider spin A, corresponding to a diagonal peak, spin A′, a one-bond neighbor of A, and spin A″, a two-bond neighbor of A. These are strongly coupled, with transfer times of <10 ms (Figs. 2, 4, 8), and therefore have nearly the same magnetization level at mixing times ≥30 ms. This is confirmed experimentally by the close match between the cross-peak (solid lines) and diagonal-peak (dashed line) intensities of the Met Cα row (Fig. 8b) at mixing times ≥30 ms. During the longer mixing time of the second experiment, the magnetization from A, A′, and A″ is transferred to more distant spins B, B′, B″, etc., resulting in a loss of intensity in the A, A′, A″ signals. This explains why a scaling factor is necessary to eliminate the reduced-intensity peaks of A–A (diagonal), A–A′ (one-bond), and A–A″ (two-bond) from the difference spectrum. Due to the fast magnetization equilibration among these three spins, the intensity ratio of the three peaks does not change much with mixing time after ~30 ms and a scaling factor that eliminates the diagonal peak A–A also removes the one-bond A–A′ and two-bond A–A″ cross peaks. A closer analysis shows that the magnetization on the initially polarized spin A is always slightly larger than that of its neighbors A′ and A″ (Fig. 8d). Thus, in a difference spectrum without one-bond cross peaks, the diagonal peaks will be slightly negative. This insight is useful for guiding the choice of the scaling factor for congested spectra, in which the one-bond cross peaks may overlap with other peaks.
Conclusion
We have shown that a T1 relaxation-compensated modification of the widely used 1H-driven 13C spin diffusion experiment allows structurally informative long-range correlation peaks to be detected in a difference spectrum. By adding a z-filter, we conduct the PDSD experiment with a constant total T1 relaxation period, and avoid the appearance of negative peaks from short-and long-range correlations of short-T1 sites in the difference spectrum. The difference between long and short mixing-time spectra, obtained using a scaling factor that makes the residual diagonal peaks slightly negative, removes all short-range cross peaks and retains only positive long-range cross peaks. The choice of the short mixing time depends on the magnetization equilibration time for the short-range correlations. For site-specifically labeled proteins, intra-residue cross peaks equilibrate by ~200 ms. Monosaccharides equilibrate their magnetization in <100 ms, and for uniformly 13C-labeled peptides and proteins, one- and two-bond cross peaks can be removed using mixing times of a few tens of milliseconds.
References
Acharya R et al (2010) Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci USA 107:15075–15080
Atalla RH, VanderHart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M (2010) Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 463:689–692
Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H (2002) Structure of a protein determined by solid-state magic-angle spinning NMR spectroscopy. Nature 420:98–102
De Paëpe G, Lewandowski JR, Loquet A, Böckmann A, Griffin RG (2008) Proton assisted recoupling and protein structure determination. J Chem Phys 129:245101
deAzevedo ER, Hu WGB, Bonagamba TJ, Schmidt-Rohr K (2000) Principles of centerband-only detection of exchange in solid-state nuclear magnetic resonance, and extension to four-time centerband-only detection of exchange. J Chem Phys 112:8988–9001
Dick-Perez M, Zhang Y, Hayes J, Salazar A, Zabotina OA, Hong M (2011) Structure and interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR. Biochemistry 50:989–1000
Higman VA et al (2009) Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR 44:245–260
Hong M (1999) Determination of multiple phi torsion angles in proteins by selective and extensive 13C labeling and two-dimensional solid-state NMR. J Magn Reson 139:389–401
Hong M (2006) Oligomeric structure, dynamics, and orientation of membrane proteins from solid-state NMR. Structure 14:1731–1740
Hong M, Jakes K (1999) Selective and extensive 13C labeling of a membrane protein for solid-state NMR investigation. J Biomol NMR 14:71–74
Hong M, Zhang Y, Hu F (2012) Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem 63:1–24
Hu F, Luo W, Hong M (2010) Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330:505–508
Lewandowski JR, De Paëpe G, Eddy MT, Struppe J, Maas W, Griffin RG (2009) Proton assisted recoupling at high spinning frequencies. J Phys Chem B 113:9062–9069
Li S, Hong M (2011) Protonation, tautomerization, and rotomeric structure of histidine: a comprehensive study by magic-angle-spinning solid-state NMR. J Am Chem Soc 133:1534–1544
Li S, Zhang Y, Hong M (2010) 3D 13C–13C–13C correlation NMR for de novo distance determination of solid proteins and application to a human alpha defensin. J Magn Reson 202:203–210
Loquet A, Lv G, Giller K, Becker S, Lange A (2011) 13C spin dilution for simplified and complete solid-state NMR resonance assignment of insoluble biological assemblies. J Am Chem Soc 133:4722–4725
Luo W, Cady SD, Hong M (2009) Immobilization of the influenza A M2 transmembrane peptide in virus envelope-mimetic lipid membranes: a solid state NMR investigation. Biochemistry 48:6361–6368
Meier BH (1994) Polarization transfer and spin diffusion in solid-state NMR. Adv Magn Opt Reson 18:1–115
Miao Y, Cross TA, Fu R (2013) Identifying inter-residue resonances in crowded 2D13C–13C chemical shift correlation spectra of membrane proteins by solid-state MAS NMR difference spectroscopy. J Biomol NMR 56:265–273
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Rienstra CM, Hohwy M, Hong M, Griffin RG (2000) 2D and 3D 15N–13C–13C NMR chemical shift correlation spectroscopy of solids: assignment of MAS spectra of peptides. J Am Chem Soc 122:10979–10990
Rienstra CM et al (2002) De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc Natl Acad Sci USA 99:10260–10265
Schmidt-Rohr K, deAzevedo ER, Bonagamba TJ (2002) Centerband-only detection of exchange (CODEX): efficient NMR analysis of slow motions in solids. In: Grant DM, Harris RK (eds) Encyclopedia of NMR. Wiley, Chichester
Takegoshi K, Nakamura S, Terao T (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett 344:631–637
Wang T, Zabotina OA, Hong M (2012) Pectin–cellulose interactions in the Arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51:9846–9856
Wang T, Park YB, Caporini MA, Rosay M, Zhong L, Cosgrove DJ, Hong M (2013) Sensitivity-enhanced solid-state NMR detection of expansin’s target in plant cell walls. Proc Natl Acad Sci USA 110:16444–16449
Acknowledgments
The amino acid and peptide component of this work was supported by NIH Grant GM088204. The plant cell wall component of this work was supported by the Center for Lignocellulose Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0001090.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tuo Wang and Jonathan K. Williams have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, T., Williams, J.K., Schmidt-Rohr, K. et al. Relaxation-compensated difference spin diffusion NMR for detecting 13C–13C long-range correlations in proteins and polysaccharides. J Biomol NMR 61, 97–107 (2015). https://doi.org/10.1007/s10858-014-9889-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-014-9889-0