
Abstract
The DREAM scheme is an efficient adiabatic homonuclear polarization-transfer method suitable for multi-dimensional experiments in biomolecular solid-state NMR. The bandwidth and dynamics of the polarization transfer in the DREAM experiment depend on a number of experimental and spin-system parameters. In order to obtain optimal results, the dependence of the cross-peak intensity on these parameters needs to be understood and carefully controlled. We introduce a simplified model to semi-quantitatively describe the polarization-transfer patterns for the relevant spin systems. Numerical simulations for all natural amino acids (except tryptophane) show the dependence of the cross-peak intensities as a function of the radio-frequency-carrier position. This dependency can be used as a guide to select the desired conditions in protein spectroscopy. Practical guidelines are given on how to set up a DREAM experiment for optimized Cα/Cβ transfer, which is important in sequential assignment experiments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Solid-state NMR is currently succeeding in addressing larger and larger protein systems. The resonance assignment of proteins with more than 200 residues is now feasible using uniformly labeled samples, with the 33 kDa C-terminal domain of the yeast prion Ure2p (285 residues) presently being the largest system with extensive de novo assignments in the solid state (Habenstein et al. 2011). Other large systems with extensive resonance assignments include HET-s(1-227)(Schuetz et al. 2010) and DsbA (223 residues)(Sperling et al. 2010; Shi et al. 2010). An important limitation is the need to acquire data showing a sufficiently high signal-to-noise ratio of the relevant cross peaks for each of the 3D spectra required for the assignment process (Schuetz et al. 2010; Habenstein et al. 2011).
It is, therefore, important to optimize the polarization-transfer efficiency of each step in order to be able to record such spectra on large systems with high cross-peak intensity. Here we concentrate on optimizing the homonuclear polarization transfer between aliphatic 13C spins using the adiabatic DREAM sequence (Verel et al. 1998, 2001). Adiabatic schemes have the advantage to transfer up to 100 % of the polarization to a certain degree independent of the magnitude of the dipolar coupling constant, while sudden schemes are limited to a maximum of 73 % (Verel et al. 1998, 2001; Hediger et al. 1995, 1994; Ernst and Meier 2010). The DREAM sequence is, therefore, extensively used as a mixing element in 3D experiments to transfer polarization mainly between Cα and Cβ (NCACB, N(CO)CACB experiments), but also between aliphatic side-chain carbons (CCC) (Gath et al. 2011; Habenstein et al. 2011; Schuetz et al. 2010). If the width of the spectrum to be recoupled is larger than one half of the spinning frequency, the DREAM sequence displays inherent frequency selectivity (Verel et al. 2001). This selectivity can be utilized to optimize the DREAM sequence for maximum transfer between selected ranges of chemical shifts. The chemical-shift selectivity of the DREAM experiment becomes particularly significant for highfield NMR (above a proton Larmor frequency of 500 MHz), and MAS frequencies between 10 and 30 kHz, typical conditions for biomolecular NMR. Fine-tuning the experimental parameters, in particular the rf-irradiation frequency, can then be used to optimize the desired transfer pathways. Optimization targets can either be an optimal compromise to observe the resonances of all amino acids, to optimize for maximum transfer in a given amino acid, or to intentionally excite only subsets of spin topologies (Radloff and Ernst 1989) to decongest crowded regions, for example, in DREAM–DREAM CCC spectra.
The strong dependence of the DREAM cross-peak patterns on the experimental conditions is exemplified by its dependence on the rf-irradiation frequency (“carrier position”) during the DREAM mixing as illustrated in Fig. 1 for the model protein ubiquitin at 24 kHz MAS. The cross-peak intensities, and sometimes even their sign vary significantly between the three spectra. Placing the carrier to the left or right side of the aliphatic region yields spectra which are approximately mirror images of each other with respect to the diagonal (Verel et al. 2001) as seen by comparing the right and leftmost panels of Fig. 1. Extracts of the valine Cβ–Cγ1/2 region (above the diagonal) are shown in Fig. 2, right column. With the given amplitude variation, the carrier position at 30 ppm is here a good choice for intense cross-peaks, while a carrier at 70 ppm leads to almost no signal. An even more pronounced dependence on the carrier position is expected for slower MAS and this is indeed clearly observed in the data recorded at 13 kHz, as shown in Fig. 2 (left column).

Overview over the aliphatic region of a series of DREAM spectra of uniformly 15N,13C labeled ubiquitin, acquired at 24 kHz MAS frequency. The carrier position during the mixing period was set to 30, 43 and 70 ppm, respectively, and is indicated by the dashed line in the spectra. Colors indicate the sign of the peak (negative, red; positive, blue). The parameters of the DREAM experiment were set as follows: center amplitude ω 1/(2π) = 11.5 kHz, difference to center amplitude at the beginning and end of the sweep ∆ = 3 kHz, estimated dipolar coupling d est = 1 kHz, mixing time τ mix = 5 ms, B 0 = 14.1 T (600 MHz)

Detail view of the valine Cγ1/2–Cβ region of a series of DREAM spectra of fully labeled ubiquitin, acquired at 13 and 24 kHz MAS frequency. The carrier position during the mixing period is indicated on the right side of the spectrum. Higher MAS frequencies lead to a more broadband transfer. DREAM parameters at 13 kHz MAS were: center amplitude ω 1/(2π) = 6 kHz, difference to center amplitude at the beginning and end of the sweep ∆ = 2 kHz, estimated dipolar coupling d est = 1 kHz. Contour levels are chosen at 10x rmsd (noise) and the spacing is a factor of 1.2
It is the scope of this work to investigate semi-quantitatively the DREAM polarization-transfer characteristics, in particular for the important Cα/Cβ transfer, for the different amino acids, and to establish guidelines how to optimize the DREAM transfer by selecting the optimum rf-irradiation frequency.
Model description
The basic principles of the DREAM experiment (see Fig. 3) are described in detail elsewhere (Verel et al. 2001; Ernst and Meier 2010). Basic insights into the cross-peak-intensity patterns of a DREAM experiment and its dependence on the experimental parameters can be gained by considering a homonuclear model spin system consisting of two or three dipolar-coupled spins. Due to the differences in the chemical-shift offsets, Ω i , of the different spins, the effective fields
will differ and the generalized HORROR condition
will be matched for different values of the rf-field amplitude for each dipolar-coupled spin pair.

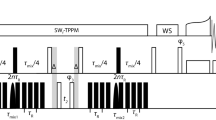
Pulse scheme of the DREAM experiment used for homonuclear 13C–13C correlation spectroscopy
For the DREAM experiment the amplitude of the rf irradiation is not fixed but is slowly varied over the experiment and is now given by ω1(T). Therefore, the resonance condition is now given by
and the HORROR condition for each of the spin pairs is matched at a different time T during the DREAM sweep. Note that, as long as the chemical-shift offsets are small compared to ω1, the solution of Eq. (3) is the classical HORROR condition: 2ω1 = ω r . For chemical-shift differences \( \left| {\Upomega_{i} - \Upomega_{j} } \right| \) smaller than the spinning frequency ω r , solutions to Eq. (3) can be found when the rf carrier is positioned in the center between the two resonances. For the example shown in Fig. 1 (24 kHz spinning frequency at 600 MHz) the maximum shift difference that can be covered is 160 ppm which includes all directly bonded 13C spin pairs. As detailed in a previous publication (Verel et al. 2001), the chemical-shift range that can be covered, such that non-selective transfer between all resonances in this range is possible, is of the order of half the spinning frequency or roughly 80 ppm, large enough to cover the aliphatic region of the spectrum. For conditions typically applied for larger proteins (Habenstein et al. 2011; Gath et al. 2011; Wasmer et al. 2009; Loquet et al. 2009), namely 850 MHz proton resonance frequency and 19 kHz MAS, the latter number is around 50 ppm, just barely enough to cover the aliphatic region of the spectrum and the choice of the carrier position becomes particularly important.
Using some simple assumptions in combination with Eq. (3), one can easily predict the expected cross-peak patterns and understand the asymmetry of the polarization transfer for a given spin system and set of experimental parameters (ω1(T) and Ω i ). The assumptions are: (i) Only spins with chemical-shift offsets within the bandwidth participate in the DREAM transfer (ii) Polarization transfer occurs only between directly bonded spins. (iii) Every polarization-transfer step is fully adiabatic with 100 % efficiency. (iv) The different transfers are fully separated in time and happen sequentially.
The consequences are most easily explored using a practical example. For this we consider a two-spin and a three-spin system with the average chemical shifts of valine from the BMRB (Cα: 62.5 ppm, Cβ: 32.7 ppm, and Cγ: 21.5 ppm) and setting the carrier at 30 ppm. For the two-spin system, we only consider the Cα and Cβ resonances. The only transition in the two-spin system is expected from Eq. (3), for a field of 600 MHz and a spinning frequency of 24 kHz, at an rf-field amplitude of 11.51 kHz. For the three-spin system, the three transitions are expected at rf-field amplitudes of 11.51 kHz (Cα/Cβ) and 11.96 kHz (Cβ/Cγ) for the strong one-bond transfers and at 11.47 kHz for the weaker (Cα/Cγ) transition (which we will neglect under assumption (ii)).
The order in which the individual recoupling conditions of individual spin pairs are met depends on the placement of the carrier and the sweep direction of the amplitude modulation. For a two-spin system (Fig. 4a), the situation is simple. At some point in the sweep (here at 11.51 kHz), the Cα ↔ Cβ resonance is met and Cα → Cβ and Cβ → Cα polarization transfer takes place and leads, assuming that the process starts from a Boltzmann populated initial state, to a symmetric spectrum, as in other types of exchange spectroscopy (Jeener et al. 1979). In a fully adiabatic process (assumption (iii)), all the intensity is found in the cross peaks, with vanishing diagonal-peak intensities (Ernst and Meier 2010). In the DREAM case, the cross peaks have the opposite sign of the diagonal peaks due to the double-quantum nature of the effective Hamiltonian.

Schematic representation of a 2D DREAM spectrum in a model 2 spin system (a) and a model 3-spin system (b). A recoupling order of the resonances of Cα → Cβ then Cβ → Cγ is assumed. The spectrum of the 3-spin system shows a strong asymmetry because different polarization transfer pathways are recoupled at different time points during the DREAM sweep. Large circles designate the peaks appearing under the assumptions given in the main text. Small circles on the diagonal indicate the source polarization
For a multi-spin system this symmetry is broken as can be seen in Fig. 4b: A rf-amplitude sweep from a low (below 11.4 kHz) to a high (above 12 kHz) amplitude leads to a sequential passage of the two resonance conditions and, as a consequence, also to a sequential recoupling of first Cα ↔ Cβ and then in a second step of Cβ ↔ Cγ. Starting on Cα, this results in a positive relayed transfer peak Cα → Cγ. The intensity of the Cα → Cβ cross peak is zero because the polarization transferred to spin Cβ in the initial Cα → Cβ transfer is relayed to Cγ in the second step. Starting on Cβ the transfer Cβ → Cα will happen first and the possible subsequent Cβ → Cγ transfer has no intensity. Starting on Cγ, only the transfer Cγ → Cβ will happen since the Cα ↔ Cβ transition has already been passed. If we release the assumption that all transfers are 100 % efficient, then also a weak Cα → Cβ cross peak will be visible as well as a weak Cβ → Cγ cross peak and weak diagonal peaks. The Cγ → Cα cross-peak cannot show up under these assumptions. These simple considerations presented above can readily be adapted to spin systems with more than three active spins to predict the pattern of cross peaks.
Numerically exact simulations
The model presented in the previous section gives insights into the pattern of the cross peaks but does not provide a quantitative picture of the polarization-transfer dynamics. In order to achieve this and gain further insight into the spin dynamics, numerical simulations of the DREAM polarization transfer were carried out for all naturally occurring amino acids, except glycine, which has no side chain, and tryptophane, whose spin system was too large for our simulations. For the simulations, the full time-dependent spin-system Hamiltonian was taken into account:
with
Here, Ω(m) and ω(m) denote the Fourier components of the chemical shift and dipolar interaction with respect to the MAS frequency. The following interactions were taken into account: (i) Isotropic chemical shifts for the amino-acids were taken from (Ye et al. 1993), for the average alpha-helical and beta-sheet values from (Wang and Jardetzky 2002); (ii) For the simulations shown, the chemical-shift anisotropy was neglected. Tests using the values from the crystalline amino acids as tabulated in ref. (Ye et al. 1993) at the highest field (850 MHz proton frequency) showed that the influence of the CSA was negligible (Westfeld 2010). (iii) Dipolar couplings were calculated from the geometry of the amino acids in the Cambridge Structural Database (Allen 2002). (iv) J couplings were set to 55 Hz for the C′–Cα, 44 Hz for the Cα–Cβ and 33 Hz for all one-bond couplings between aliphatic carbons. More-than-one-bond J-couplings were neglected. In Fig. 5a, b the results of the numerical simulations covering the aliphatic region of the spectrum are shown for a magnetic field strength corresponding to 850 MHz proton resonance frequency and spinning frequencies of 19 and 60 kHz, respectively. These two values are typical for biomolecular applications and for what the current literature calls “ultra-fast” spinning. The results of the simulations at a proton resonance frequency of 600 MHz and 13 and 24 kHz MAS, respectively, are given in Fig. 6a, b.

Simulated transfer efficiency of the DREAM scheme on a collection of amino acids as a function of the carrier position. Parameters for the simulation were a) ω r /(2π) = 19 kHz, ω1/(2π) = 9.5 kHz, ∆ = 4 kHz, d est = 1 kHz, B 0 = 19.8 T, τmix = 4 ms and b) ωr/(2π) = 60 kHz, ω1/(2π) = 30 kHz, ∆ = 18 kHz, d est = 1 kHz, B 0 = 19.8 T, τmix = 4 ms

Simulated transfer efficiency of the DREAM scheme on a collection of amino acids as a function of the carrier position. Parameters for the simulation were a ωr/(2π) = 13 kHz, ω1/(2π) = 6 kHz, ∆ = 3 kHz, d est = 1 kHz, B 0 = 14.1 T, τmix = 4 ms, and b ωr/(2π) = 24 kHz, ω1/(2π) = 11 kHz, ∆ = 4 kHz, d est = 1 kHz, B 0 = 14.1 T, τmix = 4 ms
Discussion
The simulated transfer efficiencies in Figs. 5 and 6 show the expected pronounced dependence on the rf-carrier position. For the discussion, we concentrate on the aliphatic region and classify the amino acids into three groups: (i) Proper two-spin systems, Ala, Cys, Ser, where Cα–Cβ represent the relevant part of the spin systems. (ii) Pseudo two-spin systems, Asn, Asp, His, Phe and Tyr, where the Cα–Cβ fragment is connected to an aromatic or side-chain carbonyl or carboxamide carbon spin. These Cγ resonances are spectrally more detuned from the Cβ and their presence plays a less decisive role. For many purposes, they can be compared to the two-spin systems of class (i). (iii) The third class are multi-spin systems, where Cα–Cβ–CX represent the active spin system–system. Here, CX refers to at least one side-chain carbon atom fully within the bandwidth of the DREAM recoupling. This group contains Arg, Gln, Glu, Ile, Leu, Lys, Met, Pro, Tyr and Val. Glycine is in this context an effective one-spin system and leads to diagonal peaks only in a two-dimensional DREAM correlation spectrum. Tryptophane was not calculated.
The transfer profiles (transfer efficiency as a function of the carrier frequency) in the group of the effective two-spin systems lead to an almost symmetric 2D spectrum (Cα → Cβ and Cβ → Cα) where cross peaks are both negative and of similar intensity, following the example of Fig. 4a. The transfer efficiency as a function of the carrier offset is approximately symmetric around the mean frequency of Cα and Cβ. The width of the profile increases with spinning frequency and decreases with increasing static magnetic-field strength. At 13 kHz/600 MHz and 19 kHz/850 MHz, a carrier frequency around 55 ppm is a good compromise for efficient transfer in all amino acids belonging to the class of effective two-spin systems. At higher spinning frequencies, the exact carrier position becomes less critical, in fact at 60 kHz, any carrier position between 10 and 70 ppm is acceptable and gives efficient polarization transfer. We have repeated these calculations which used the amino-acid shifts using typical α-helical or β-sheet values for the isotropic chemical shifts in a protein while keeping all other parameters constant, and found only minor changes in the transfer profiles. We note that many of the transfer curves in Figs. 5 and 6 show a “dip” for the carrier positioned in the center between the Cα and Cβ resonances. Note that in these Figures, the Cα and Cβ resonance positions are indicated by solid and dashed vertical lines. These dips vanish when the simulations are done neglecting the homonuclear J-coupling Hamiltonian (Westfeld 2010). The dip is caused by J-coupling mediated zero-quantum polarization transfer which destructively interferes with the double-quantum HORROR polarization transfer. Similar effects were observed for offset-compensated HORROR versions (Verel and Meier 2004).
For the class of Cα–Cβ–CX multi-spin systems, the effect of the carrier position is more complex (Fig. 7). The width of the profile for Cα → Cβ transfer is reduced compared to the two-spin group, and the spectra can become very asymmetric as in the example shown in Fig. 7b. For carrier frequencies above about 40 ppm, the negative Cα → Cβ peaks (for most amino acids below the diagonal) are significantly more intense than the Cβ → Cα peaks. For carrier positions below 40 ppm, the negative Cβ → Cα peaks is significantly more intense than the Cα → Cβ peaks leading to spectra with more intensity below (or above) the diagonal. This tendency is clearly also observed in Fig. 1 and will be further compared to the experiments below. Note that these observations are made for a DREAM sweep with increasing amplitude. For a sweep with decreasing amplitude, the spectra will be mirrored at the diagonal. The reason for the different recoupling bandwidths of the Cα/Cβ transfer is due to the presence or absence of a third spin to which a relay transfer at the expense of a direct transfer is possible as also illustrated in Fig. 4.

Cross-peak patterns for valine in ubiquitin. (a) Simulation of the transfer efficiency for the different spin pairs in the spin system. The three dashed vertical lines indicate the three carrier positions used in the experiments. Extracts from DREAM spectra recorded on ubiquitin (Fig. 1) using a carrier setting of 30 ppm (b), 43 ppm (c) and 70 ppm (d). For better comparability the spectra in the right column are shown with swapped direct and indirect frequency axes. Simulations were done and spectra recorded at 24 kHz MAS frequency and at a proton resonance frequency of 600 MHz. Experimental parameters are given in the caption of Fig. 1
Figure 7 compares the calculated transfer efficiencies for the Cα–Cβ–CX four-spin system of valine at three carrier positions with experimental intensities from the ubiquitin spectra (Fig. 1) for the Cα → Cβ, Cα → Cγ1 and Cα → Cγ2 und the inverse cross peaks. For a carrier position at 30 ppm we predict (Fig. 7a) that the strongest cross peaks are the Cβ → Cα peaks (negative). The Cα → Cγ (both Cγ1 and Cγ2) cross peak intensities are significant but weaker and positive and we expect weak negative cross peaks for Cα → Cβ and Cγ → Cα. This pattern is indeed observed experimentally as shown in Fig. 7b. For a carrier position at 43 ppm, no very strong cross peaks are predicted, with the strongest ones being Cα → Cβ and Cβ → Cα (both negative) followed by Cγ → Cα (positive). Again, the experimental intensities follow the prediction (Fig. 7c). For a carrier position at 70 ppm, the strongest cross peaks predicted are Cβ → Cα (negative) followed by Cγ → Cα (positive) and the predicted behavior is observed experimentally (Fig. 7d). We note that the Cα → Cγ (or Cγ → Cα) peaks can be positive or negative. The positive peaks (typically significantly stronger) are caused by a Cα → Cβ → Cγ relayed transfer, the negative peaks by a direct Cα → Cγ transfer by the (smaller) direct dipolar coupling between Cα and Cγ. The sign of the different cross peaks is a hallmark of the topology of the spin system and can be used for assignment purposes.
Summary and conclusions
We have described a simple model for understanding semi-quantitatively the cross-peak intensity in DREAM spectra of the different natural amino acids. The simple rules and in particular the numerical simulations provide important information how to set up DREAM experiments on proteins in order to obtain the desired polarization transfers. In the following, we summarize the conclusions for a prototypical experiment where optimal Cα–Cβ transfer is requested for a maximum number of amino-acid residues (e.g. in the context of an NCACB experiment) at 850 MHz proton resonance frequency and 19 kHz MAS: (i) The carrier should be placed at around 57 ppm. (ii) The HORROR condition is lowered in the case of the chemical-shift offsets: For a chemical-shift difference of 30 ppm between the Cα and Cβ resonances (e.g. valine), the HORROR condition appears at 8.5 kHz rf-field amplitude instead of at 9.5 kHz. The estimated dipolar coupling d est used for calculating the exact shapes (Verel 2001; Ernst and Meier 2010) is set to 1 kHz corresponding to a typical recoupled 13C–13C coupling constant. (iii) Faster MAS frequencies will lead to a more broadband recoupling, as stronger radio-frequency fields have to be used. However, for a Cα/Cβ transfers, MAS frequencies of 15 kHz at a static field of 19.8 T are fast enough to generate intense cross peaks. (iv) The duration of the DREAM sweep is optimized to one-bond dipolar couplings, and values in the range of 3–6 ms were found to be adequate for 13C–13C transfer.
These principles allow the optimization of the DREAM condition for intense broadband Cα/Cβ transfer. This transfer is essential in multi-dimensional assignment spectra for larger proteins, where the use of the Cβ spins is central to achieve better spectral dispersion. More generally, the same is also true for the three-dimensional NCACB experiment, which we extensively use as the most sensitive experiment to obtain a 3D spectral fingerprint for larger proteins. In 3D CCC experiments, fine-tuning of the offset frequencies and amplitude offsets should allow spectral editing to alleviate overlap, important in the context of the study of large molecules.
Experimental
Simulation of the DREAM transfer
Simulations were programmed in C++ using the GAMMA spin-simulation framework (Smith et al. 1994). All simulations are powder averages over 100 crystallite orientations (Cheng et al. 1973). The time dependence of the Hamiltonian was approximated by subdividing each rotor period into 250 time steps with a time-constant Hamiltonian. The DREAM sweep was approximated by sampling the amplitude modulation with 200 discrete amplitude steps. For the simulations atom coordinates of crystalline single amino acids from the Cambridge Structural Database were used. The following records were used for the amino acids: LALNIN, ARGHCL10, ADSPAM2, ASPART10, LCYSTN04, LGLUTA, GLUTAM03, GLYHCL, DLHIST, DALILU10, FEQYUW, LYSCLH02, METHCL, PHALNC01, DLPROL, LSERMH10, LTHREO03, LTYRHC10, VALEHC11.
NMR spectroscopy
DREAM spectra of U-(13C, 15N) Ubiquitin were acquired on a Bruker AV600 spectrometer operating at a static field of 14.09 T corresponding to a proton Larmor frequency of 600 MHz. A Bruker 3.2 mm triple-resonance probe was used. MAS was actively stabilized to within ±10 Hz. The temperature of the sample was stabilized at about 3°C using a Bruker VT controller in combination with a BCU-Xtreme cooling unit. In spectra recorded at 13 kHz MAS the SPINAL64 scheme (Fung et al. 2000) and in spectra at 24 kHz MAS the XiX scheme (Detken et al. 2002) was used during t 1 and t 2 with an rf-field strength of 100 kHz. During the DREAM mixing cw-decoupling with a field strength of 100 kHz was applied. The tangential amplitude modulation of the DREAM sweep was approximated with 1,000 discrete steps. The mixing time was set to 5 ms. In the spectrum recorded at 24 kHz MAS the center amplitude of the DREAM sweep was 11.5 kHz, the initial Δ of the sweep 3 kHz and the estimated dipolar coupling 1 kHz. In the spectrum recorded at 13 kHz MAS the center amplitude was 6 kHz, the initial Δ 2 kHz and the estimated dipolar coupling 1 kHz. In the indirect dimension 1,024 points with 16 scans each were acquired using TPPI (Marion and Wüthrich 1983) for sign discrimination. The total measurement time was 9 h per spectrum.
Spectra were processed with nmrPipe (Delaglio et al. 1995) using a squared cosine apodization function in both dimensions. Peaks were picked using Sparky (Kneller and Kuntz 1993) and resonance lists were evaluated using custom Perl scripts.
Abbreviations
- NMR:
-
Nuclear magnetic resonance
- MAS:
-
Magic-angle spinning
- CP:
-
Cross-polarization
- DREAM:
-
Dipolar recoupling enhanced by amplitude modulation
References
Allen FH (2002) The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr B 58(Pt 3 Pt 1):380–388
Cheng VB, Suzukawa HH, Wolfsberg M (1973) Investigations of a nonrandom numerical-method for multidimensional integration. J Chem Phys 59(8):3992–3999
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMR-Pipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Detken A, Hardy E, Ernst M, Meier BH (2002) Simple and efficient decoupling in magic-angle spinning solid- state NMR: the XiX scheme. Chem Phys Lett 356(3–4):298–304
Ernst M, Meier BH (2010) Adiabatic polarization-transfer methods in MAS spectroscopy. Encycl Magn Reson (EMR) online: doi:10.1002/9780470034590.emrstm9780470030004.pub9780470034592
Fung B, Khitrin A, Ermolaev K (2000) An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson 142(1):97–101
Gath J, Habenstein B, Bousset L, Melki R, Meier BH, Böckmann A (2011) Solid-state NMR sequential assignments of α-synuclein. Biomol NMR Assign: (in press, available on the web)
Habenstein B, Wasmer C, Bousset L, Sourigues Y, Schütz A, Loquet A, Meier BH, Melki R, Böckmann A (2011) Extensive de novo solid-state NMR assignments of the 33 kDa C-terminal domain of the Ure2 prion. J Biomol NMR 51(3):235–243
Hediger S, Meier BH, KURUR N, BODENHAUSEN G, Ernst R (1994) NMR cross polarization by adiabatic passage through the Hartmann-Hahn condition (APHH). Chem Phys Lett 223:283–288
Hediger S, Meier BH, Ernst R (1995) Adiabatic passage Hartmann-Hahn cross polarization in NMR under magic angle sample spinning. Chem Phys Lett 240:449
Jeener J, Meier BH, Bachmann P, Ernst RR (1979) Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys 71(11):4546
Kneller D, Kuntz I (1993) UCSF sparky—an NMR display, annotation and assignment tool. J Cell Biochem 53:254
Loquet A, Bousset L, Gardiennet C, Sourigues Y, Wasmer C, Habenstein B, Schütz A, Meier BH, Melki R, Böckmann A (2009) Prion fibrils of Ure2p assembled under physiological conditions contain highly ordered, natively folded modules. J Mol Biol 394(1):108–118
Marion D, Wüthrich K (1983) Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem Biophys Res Commun 113:967–974
Radloff C, Ernst R (1989) Spin topology filtration in NMR. Mol Phys 66(1):161–197
Schuetz A, Wasmer C, Habenstein B, Verel R, Greenwald J, Riek R, Böckmann A, Meier BH (2010) Protocols for the sequential solid-state NMR spectroscopic assignment of a uniformly labeled 25 kDa protein: HET-s(1-227). ChemBioChem 11(11):1543–1551
Shi L, Kawamura I, Jung K-H, Brown LS, Ladizhansky V (2010) Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew Chem Int Ed Engl 50(6):1302–1305
Smith S, Levante T, Meier BH, Ernst R (1994) Computer simulations in magnetic resonance: an object oriented programming approach. J Magn Reson Ser A 106:75–105
Sperling LJ, Berthold DA, Sasser TL, Jeisy-Scott V, Rienstra CM (2010) Assignment strategies for large proteins by magic-angle spinning NMR: the 21-kDa disulfide-bond-forming enzyme DsbA. J Mol Biol 399(2):268–282
Verel R (2001) Adiabatic methods for homonuclear dipolar recoupling in magic angle spinning solid-state NMR. DISS ETH 14152
Verel R, Meier BH (2004) Polarization-transfer methods in solid-state magic-angle- spinning NMR: adiabatic CN pulse sequences. ChemPhysChem 5(6):851–862
Verel R, Baldus M, Ernst M, Meier BH (1998) A homonuclear spin-pair filter for solid-state NMR based on adiabatic-passage techniques. Chem Phys Lett 287:421–428
Verel R, Ernst M, Meier BH (2001) Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J Magn Reson 150(1):81–99
Wang Y, Jardetzky O (2002) Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci 11(4):852–861
Wasmer C, Schütz A, Loquet A, Buhtz C, Greenwald J, Riek R, Böckmann A, Meier BH (2009) The molecular organization of the fungal prion HET-s in its amyloid form. J Mol Biol 394(1):119–127
Westfeld T (2010) Polarization transfer dynamics in multi-spin systems using the DREAM scheme. Diss ETH 19196:1–153
Ye C, Fu R, Hu J, HOU L, Ding S (1993) C-13 chemical-shift anisotropies of solid amino-acids. Magn Reson Chem 31(8):699–704
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Westfeld, T., Verel, R., Ernst, M. et al. Properties of the DREAM scheme and its optimization for application to proteins. J Biomol NMR 53, 103–112 (2012). https://doi.org/10.1007/s10858-012-9627-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-012-9627-4