
Abstract
We present a comprehensive analysis of protein dynamics for a micro-crystallin protein in the solid-state. Experimental data include 15N T 1 relaxation times measured at two different magnetic fields as well as 1H–15N dipole, 15N CSA cross correlated relaxation rates which are sensitive to the spectral density function J(0) and are thus a measure of T 2 in the solid-state. In addition, global order parameters are included from a 1H,15N dipolar recoupling experiment. The data are analyzed within the framework of the extended model-free Clore–Lipari–Szabo theory. We find slow motional correlation times in the range of 5 and 150 ns. Assuming a wobbling in a cone motion, the amplitude of motion of the respective amide moiety is on the order of 10° for the half-opening angle of the cone in most of the cases. The experiments are demonstrated using a perdeuterated sample of the chicken α-spectrin SH3 domain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Characterization of internal protein dynamics is important to obtain a better understanding of the energetics involved in ligand binding (Frederick et al. 2007; Lange et al. 2008), the enzymatic activity of proteins (Eisenmesser et al. 2002; Henzler-Wildman and Kern 2007), protein folding and unfolding (Grey et al. 2006; Korzhnev et al. 2004; Vallurupalli et al. 2008), and to get insight into the dynamics of ion binding sites in RNA and proteins (Eichmüller and Skrynnikov 2005, 2007; Hoogstraten et al. 2000).
Solution-state NMR relaxation measurements are restricted to the detection of motional time scales faster than the tumbling correlation time τ R of the protein under investigation (Cavanagh et al. 1996). Motion slower than the overall correlation time is masked as the respective spectral density functions decay rapidly above τ R . Dynamics on a μs to ms timescale can be probed using R 1ρ type sequences. The time domain inbetween (ns–μs) is not easily accessibly and can only be sampled by artificially increasing the overall motional correlation time by extending the size of the molecule (Zhang et al. 2006), or by increasing the viscosity of the solution (Xu et al. 2009; Zeeb et al. 2003), assuming that local dynamics are unaffected. Alternatively, slow motional processes can be identified by comparison of order parameters obtained from 15N relaxation and RDC measurements using multiple alignment media (Bouvignies et al. 2005; Lakomek et al. 2005). RDC measurements will, however, only yield information on the amplitude of the implicated dynamics. The timescale of motion can be accessed only indirectly by exclusion.
On the other hand, analysis of motion comes more and more into the focus of MAS solid-state NMR spectroscopy. In the past, e.g. the loop motion in triosephosphate isomerase was characterized (Williams and McDermott 1995). Analysis of dynamics in the solid-state is even more so of interest as certain parts of the protein are apparently mobile, as the cross-polarization transfer dynamics for resonances in mobile regions of the protein can be vanishing (Andronesi et al. 2005; Helmus et al. 2008; Wasmer et al. 2008). In this case, a complete set of resonances can only be employed when scalar transfer sequences are employed (Agarwal and Reif 2008). In contrast to solution-state, overall tumbling is absent in the solid-state. In an immobilized sample, relaxation is exclusively resulting from local structural fluctuations. The dynamic properties of a protein should therefore be accessible with very high accuracy. In particular, motion beyond the overall correlation time limit, imposed in the analysis of dynamics relying on solution-state NMR relaxation data, should be within reach. 15N-T 1 (Chevelkov et al. 2008; Cole and Torchia 1991; Giraud et al. 2004, 2005, 2007), heteronuclear Overhauser effects (Giraud et al. 2006) and 15N-CSA, 1H–15N dipole cross correlated relaxation (Chevelkov et al. 2007a; Skrynnikov 2007) in uniformly isotopically enriched proteins can nowadays be measured routinely on uniformly isotopically enriched proteins. Site-specific order parameters were obtained from dipolar recoupling MAS solid-state NMR experiments for various backbone and sidechain 13C–1H moieties (Franks et al. 2005; Huster et al. 2001; Lorieau and McDermott 2006; Lorieau et al. 2008; Sackewitz et al. 2008). Side chain dynamic information is accessible making use of the deuterium quadrupolar interaction by interpretation of the spinning sideband manifold (Hologne et al. 2005, 2006a, b). 2H/13C-T 1 methyl relaxation measurements allow a more detailed characterization of the motional time scale detected in order parameter experiments (Agarwal et al. 2008; Reif et al. 2006). A comparison of relaxation data obtained in the solid-state and in solution reveals that dynamics in both states is highly similar (Agarwal et al. 2008; Chevelkov et al. 2007c). This opens perspectives for a combined analysis of motion using both solution and solid-state NMR spectroscopy (Palmer-III et al. 1996).
Recently, we suggested a labeling scheme which is based on high levels of deuteration to eliminate most of the undesired proton–proton dipolar interactions (Chevelkov et al. 2006). The perdeuterated protein is recrystallized from a buffer containing 90% D2O in order to suppress anisotropic interactions among exchangeable sites. Given the fact that high power heteronuclear and homonuclear decoupling is not required, the temperature of the sample is well defined and dynamic properties can be quantified with high accuracy. The deuteration scheme enables a spin-diffusion free determination of dynamic parameters. The goal of the paper is to integrate 15N-T 1, 1H–15N dipole, 15N CSA cross correlated relaxation and 1H–15N dipolar coupling measurements performed in our laboratory to yield a quantitative description of dynamics in the solid-state. The experiments are carried out using a perdeuterated sample of the chicken α-spectrin SH3 domain (Fig. 1).

Materials and methods
Sample preparation
A pET3d derivative coding for α-spectrin SH3 domain from chicken brain was a gift of M. Saraste. Protein was expressed in E. coli BL21 (DE3) in M9 minimal medium with 100% D2O with 4 g/L 2H8-glycerol as the sole carbon source, together with 1 g/L 15N–NH4Cl. Cells were grown at 37°C up to an optical density (OD600 nm) of 0.6. The temperature was then decreased to 22°C and induction was started with 1 mM IPTG overnight. Purification of the cell extract was carried out in H2O (anion exchange on a Q-Sepharose FF column, followed by gel filtration on a Superdex75 column; Pauli et al. 2000). 10 mg of the purified protein was lyophilized and redissolved in H2O/D2O using a mixing ratio of 10:90 with respect to solvent exchangeable protons. Microcrystalline precipitates were obtained by mixing the protein solution (10 mg/mL) at a ratio of 1:1 (v/v) with a 200 mM (NH4)2SO4 solution containing 90% D2O. The pH value was adjusted to around seven by exposing the sample to an alkaline atmosphere, monitoring the pH with pH sensitive paper strips.
NMR spectroscopy
Measurements of the 15N-T 1 relaxation time (Chevelkov et al. 2008), the 1H–15N dipole, 15N CSA cross correlated relaxation rate η DD/CSA (Chevelkov et al. 2007a, b; Chevelkov and Reif 2008) and 1H–15N dipolar recoupling experiments (Chevelkov et al. 2009) were carried out as described previously. In particular, 1H detection in combination with perdeuteration was employed to achieve high sensitivity and resolution (Chevelkov et al. 2003; Reif et al. 2001; Reif and Griffin 2003). All experiments are carried out at an effective sample temperature of 11°C, using a perdeuterated, 15N labeled sample of the α-spectrin SH3 which was recrystallized using a mixture of 10% H2O and 90% D2O in the crystallization buffer, as described in (Chevelkov et al. 2006). The assignment of the resonances was obtained using HNCACB type experiments (Linser et al. 2008).
Theoretical background
The analysis of motion in the solid-state is carried out in the framework of Redfield (1957) theory. This approach seems to be valid as we are only considering motional time scales which are faster compared to the size of the involved anisotropic interactions.
The experiment employed to measure 15N T 1 is described in detail in reference (Chevelkov et al. 2008). The measured relaxation rate R 1(15N) is related to the size of the N–H dipolar coupling d, the chemical shift anisotropy c and the spectral densitiy function J(ω) according to (Cavanagh et al. 1996; Torchia and Szabo 1982)
with
and r NH refers to the 1H–15N bond length. ωH and ωN represent the 1H and the 15N Larmor frequencies, respectively. The frequency of the 1H,15N dipole–dipole interaction is denoted as ωHN. The 15N CSA can be assumed to be axially symmetric. The 15N–1H bond is tilted by ~20° with respect to the principal axis of the 15N CSA tensor (Chekmenev et al. 2004). Typical values for the anisotropy of the 15N chemical shift are \( \Updelta \sigma = \sigma_{||} - \sigma_{ \bot } = 170 \pm 8\,{\text{ppm}} \), or σ z = 106 ± 6 ppm (Chekmenev et al. 2004; Franks et al. 2005; Hall and Fushman 2006; Wylie et al. 2006, 2007). In the absence of motion, an effective N–H bond length of r NH = (<r 3NH >)1/3 = 1.015 Å is assumed (Yao et al. 2008).
In the solid-state, the exact form of the spectral density function depends on the underlying motional model (Torchia and Szabo 1982). In the framework of the extended model free formalism (Clore et al. 1990; Lipari and Szabo 1982), the spectral density functions J m(ω) are expressed as lorentzian functions depending on two correlation times τ s and τ F , and two order parameters S S and S F , respectively, referring to slow and fast motional processes
In order to obtain a “T 2 type” observable which depends on J(0), 1H–15N dipole, 15N CSA cross correlated relaxation rates η DD/CSA were measured. Quantitative values for η DD/CSA were extracted from a constant-time experiment in which protons are not decoupled in the indirect evolution period (Chevelkov et al. 2007a). The integral intensity ratio of the N–Hα/N–Hβ multiplet components after a relaxation delay is directly related to the cross correlated relaxation rates η DD/CSA. In the solid-state, the Hamiltonian describing an isolated 1H,15N spin pair is inhomogeneous in the sense of Maricq and Waugh (Maricq and Waugh 1979). If the spinning rate is larger than the size of the CSA and dipole-dipole interaction, the 15N line width is only determined by motional processes. However, even at a MAS rotation frequency of 24 kHz coherent effects are not completely averaged out (Chevelkov et al. 2007b). In a spin system, in which the proton of the 1H,15N pair is integrated into a proton spin network, the Hamiltonian is no longer inhomogeneous. Similar arguments apply for protonated samples in the limit of insufficient 1H decoupling. The decay of 15N transverse magnetization is therefore not only due to relaxation, but influenced as well by the proton spin bath. In this system, the 15N T 2 relaxation can not be used directly to probe motional properties of the protein. Even in the heavily proton dilute protein sample used in this study, the proton line width depends still weakly on the MAS frequency (Chevelkov et al. 2006), reflecting residual long range proton interactions. Simulations in which three additional protons are taken into account (assuming mutual 1H, 1H dipolar interactions on the order of 800–1,200 Hz) yield a uniform increase in the line width of both the Hα and Hβ multiplet component of the 15N spectrum (Chevelkov et al. 2007a). Our experimental approach is supported by numerical simulations in which coherent and incoherent cross correlation effects are explicitly coded under magic angle spinning (Skrynnikov 2007). Incoherent effects like cross correlated relaxation result in a differential line width of the N–Hα/N–Hβ multiplet components. Coherent effects like static correlations between the dipolar and the CSA interaction will at the same time not affect the relative N–Hα/N–Hβ resonance line width.
The cross correlated relaxation rate η DD/CSA can be described again as a function of spectral density functions, using the equation (Fushman and Cowburn 1998; Tjandra et al. 1996)
where P 2(x) = (3x 2 − 1)/2 corresponds to the second order Legendre polynomial, θ represents the angle between the principal axis of the 15N–1H dipolar vector and the 15N CSA shielding tensor, and adopts values on the order of ca. 20°.
The last input parameter employed in the fitting analysis is the generalized order parameter S = S S S F . In solid-state NMR, S refers to the ratio of the motionally averaged dipolar coupling to its value in the static limit. The generalized order parameter S for each amide moiety is therefore implicitly contained in the size of the individual experimental N–H dipolar coupling. To quantitatively access the dipolar couplings, we made use of a phase-inverted CP (CPPI) experiment which is insensitive to RF inhomogeneity and thus yields the absolute dipolar coupling with high accuracy (Dvinskikh et al. 2003, 2005; Wu and Zilm 1993). We have shown that fluctuations of the N–H bond length due to differential hydrogen bonding are within the experimental error of the estimation of the size of the coupling and can therefore be neglected in the analysis (Chevelkov et al. 2009). In case the motional model implies a wobbling of the N–H bond vector within a cone with an half-opening angle α 0, the general order parameter S can be expressed as (Lipari and Szabo 1981; Palmer-III et al. 1996)
In this model, an half-opening angle of α = 21° yields an general order parameter S = 0.9.
Results and discussion
In the following, we combine all experimental results (15 R 1 measured at an external field of 14.1 T and 21.1 T, corresponding to 1H Larmor frequency of 600 MHz and 900 MHz; 1H–15N dipole, 15N CSA cross correlated relaxation rate η DD/CSA and 1H,15N dipolar couplings) in a multi-dimensional grid search to find the best fit conditions in the framework of an extended model-free analysis (Clore et al. 1990; Lipari and Szabo 1982). In total, the data contains four experimental observables. The extended model-free spectral densities are dependent on four parameters, in particular, the slow and fast motional correlation times τ s and τ F , and the order parameters for slow and fast motion S S and S F , respectively. Obviously, the data are rather sparse. Therefore, the results obtained for each amide need to be evaluated to rationalize if the data and the employed model allow a consistent description of the system. The root mean square deviation χ between experimental and theoretical rates is defined as
Superscripts theo and expt denote the theoretical and experimental values for the 15N longitudinal relaxation rate R 1 and the 1H–15N dipole, 15N CSA cross-correlated relaxation rate η DD/CSA. The theoretical values for the relaxation rates were calculated using Eqs. 1–4. In all calculations, the dipolar coupling derived general order parameter is employed. A 3D grid search was performed allowing the parameters times τ S , τ F and S 2 S to float freely, while the order parameter of fast motion S 2 F was calculated according to S F = S/S S .
Figure 2 shows RMSD contour plots for residue Q16 of the α-spectrin SH3 domain as a function of τ S and S 2 S . τ F was set to 22 ps, which corresponds to its optimal value obtained in the course of the grid search. Whereas in the fitting underlying Fig. 2a only 15N-T 1 are included, Fig. 2b–d contain as well cross-correlated relaxation data (η DD/CSA). We find that the minimum for the fit of the motional correlation time τ S is more restricted if η DD/CSA is taken into account. Inclusion of an additional 15N-T 1 relaxation time measured at a different external field strength increases the steepness of the minimum, but leaves the best fit for τ S and S 2 S approximately unaltered. This is in agreement with previous findings (Chevelkov et al. 2007c; Giraud et al. 2005).

Rms difference plots between experimental and theoretical data as a function of S 2 S and τ S for the residue Q16 in α-spectrin SH3. For the best fit, we obtain τ F = 22 ps, and S 2 F = 0.819. Data included in the fit are: a 15N T 1 measured at 14.1 T and 21.1 T. b 15N T 1 measured at 14.1 T and η DD/CSA. c 15N T 1 measured at 21.1 T +η DD/CSA. d 15N T 1 measured at 14.1 T, 15N T 1 measured at 21.1 T and η DD/CSA
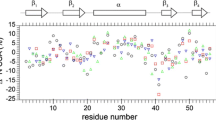
In the following, we represent the various best fit values as a function of the protein sequence. Figure 3 shows the overall order parameter as well as the order parameter for slow motional processes, together with the implicated half-opening angle, for the α-spectrin SH3 domain. The general order parameter <S> was set to 1 assuming an effective N–H bond length of r eff = (<r −3NH >)−1/3 = 1.015 Å (Yao et al. 2008). We find in general lower order parameters at the N- and C-terminus. The average order parameter for slow motional processes in on the order of S 2 S = 0.95, corresponding to an half-opening angle of ca. 10° assuming a diffusion in a cone motion (Lipari and Szabo 1981).

Order parameter S 2 as a function of the primary sequence in α-spectrin SH3. Black squares denote the order parameter of slow motional processes, whereas red circles indicate the overall order parameter S 2 S S 2 F . The vertical axis on the right hand side of the figure displays the half-opening angle α0 assuming a “wobbling in a cone” motion, making use of Eq. 5
The correlation times of slow and fast motion τ S and τ F are shown in Fig. 4. Black squares represent the best fit values. Red circles and green triangles denote the result of the RMSD fitting obtained by changing the experimental 15N-T 1 relaxation time (a, b), the measured 1H–15N, 15N-CSA cross correlated relaxation rate η DD/CSA (c, d) and the overall motional parameter 1 − <S> (e, f) by ± 10%, respectively. Residues displayed at the bottom of Fig. 4 show unusual large values and are therefore plotted separately. Non-canonical residues are L8, Y13, R21, D40, V46, R49 and D62. For residues Y13, R21 and D40, we find very high slow motional order parameters S 2 S which are on the order of 0.9959, 0.9871 and 0.9932, respectively. Motion for those residues seems considerably restricted. The high value for S 2 S implies that only fast motional processes contribute to the spectral density (2). For canonical residues, the slow motional correlation time τ S varies between 5 and 70 ns. The second set of non-canonical residues comprises residues L8, V46, R49 and D62. For those residues, τ F fits to values on the order of several nanoseconds and τ S can be as large as 150 ns. For these residues, we find high values of η DD/CSA on the order of 10 Hz, or larger. Residue V46 is located in the distal loop. The adjecent residues N47 and D48 have strongly increased B-factors in the X-ray structure and are not observed in solid-state NMR spectra. It is therefore plausible that V46 is undergoing slow dynamics. This is supported by the exceptionally long value for τ F (4.5 ns) for V46 and R49. For these residues, the extended model-free analysis might represent an over-simplification. One more motional mode seems to be required in order to describe appropriately the underlying dynamics for these residues, as one would expect a vibrational mode as the fastest dynamic process. Fitting the data using a simple Lipari–Szabo analysis with only two unknown parameters, namely one correlation time τ and one order parameter S 2, reproduces the slow motional correlation time in general rather well (data not shown). We therefore believe that the indicated values for τ S for the amide moieties of V46 and R49 correctly describe this parameter. A similar explanation applies to L8 and D62 which are located at the N- and C-terminus of the protein and which are virtually the first and last residue for which an electron density can be assigned. D40 is contained in the n-Scr loop. It is noteworthy that the fitting analysis cannot be carried out for residues S36, T37, N38 and K39, as these residues yield significantly reduced intensities in 1H,15N correlation spectra. The achieveable signal-to-noise ratio for those residues is insufficient to allow a quantitative extraction of any of the described relaxation rates. The reduced intensity might be indicative for very slow motion or a chemical exchange process on a μs–ms time scale. In general, we find τ F to be on the order of 5–50 ps which would be expected for vibrational dynamics. This is in agreement with the results obtained by Mack et al. (2000) who used field dependent bulk 2H T 1 relaxation times and quadrupolar order parameters of exchangeable deuterons in RNase H to quantify backbone motion. Variations in the experimental 15N-T 1 relaxation time imposes the largest error on the extracted motional correlation times τ S and τ F . The fitting procedure breaks down and yields a very larger error, in case the artificially increased 15N-T 1 relaxation time at 14.1 T (red circles in Fig. 4) becomes close to, or larger than the experimental 15N-T 1 relaxation time measured at 21.1 T. Variations in the cross correlated relaxation rate η DD/CSA and 1H–15N order parameters produce smaller variations in the extracted slow and fast motional correlation time τ S and τ F (Fig. 4c–f).


Slow and fast motional correlation times τ S and τ F as a function of the primary sequence in α-spectrin SH3. Black squares refer to the best fit value for τ S and τ F , respectively. Red circles and green triangles denote the result of the RMSD fitting obtained by changing the experimental 15N-T 1 relaxation time obtained at 14.1 T (a, b), the measured 1H–15N, 15N-CSA cross correlated relaxation rate η DD/CSA (c, d) and the overall motional parameter 1 − <S> (e, f) by +/– 10%, respectively. The panels at the bottom of each figure show the results obtained for Y13, R21, D40 and L8, V46, R49, D62, respectively, for which we find particularly long values for τ S . For residues L8, V46, R49 and D62, τ F becomes exceptionally long, indicating that motion is more complicated, and that the extended model free analysis is no longer sufficient to describe the motional process
So far, we are not able to detect long range correlated dynamics which was suggested by the alternating pattern which we observed previously in the 15N-T 1 relaxation times in β-sheet β2 comprising residues 30–35 (Chevelkov et al. 2008). Long range collective motion was suggested recently for ubiquitin (Lakomek et al. 2005) and protein G (Bouvignies et al. 2005). At this point, we cannot pursue this question in greater detail as the data—in particular in this part of the protein—is still rather sparse. In total, we were able to analyze 34 out of 53 possible amide moieties. Work is going on in our laboratory to obtain a more complete set of data.
Conclusion
In conclusion, we have shown that the dynamic properties of the protein backbone can be characterized in the solid-state with high accuracy, using 15N-T 1 relaxation times measured at two different fields, 1H–15N, 15N-CSA cross correlated relaxation rate η DD/CSA and 1H–15N dipolar coupling measurements to obtain overall order parameters. Using the micro-crystalline α-spectrin SH3 domain as a model system, we find slow motional correlation times between 5 and 150 ns. Assuming a wobbling in a cone motion, the amplitude of motion of the respective amide moiety is on the order of 10° for the half-opening angle of the cone in most of the cases. We expect that these experiments will find widespread application in the characterization of dynamic processes of biomolecules such as membrane proteins and amyloidogenic peptides and proteins.
References
Agarwal V, Reif B (2008) Residual methyl protonation in perdeuterated proteins for multidimensional correlation experiments in MAS solid-state NMR spectroscopy. J Magn Reson 194:16–24
Agarwal V, Xue Y, Skrynnikov NR, Reif B (2008) Protein side-chain dynamics as observed by solution- and solid-state NMR: a similarity revealed. J Am Chem Soc 130:16611–16621
Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M (2005) Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J Am Chem Soc 127:12965–12974
Bouvignies G, Bernado P, Meier S, Cho K, Grzesiek S, Bruschweiler R, Blackledge M (2005) Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc Natl Acad Sci USA 102:13885–13890
Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ (1996) Protein NMR spectroscopy: principles and practice. Academic Press, San Diego
Chekmenev EY, Zhang Q, Waddell KW, Mashuta MS, Wittebort RJ (2004) 15N chemical shielding in glycyl tripeptides: measurement by solid-state NMR and correlation with X-ray structure. J Am Chem Soc 126:379–384
Chevelkov V, Reif B (2008) TROSY effects in MAS solid-state NMR. Concepts NMR 32A:143–156
Chevelkov V, van Rossum BJ, Castellani F, Rehbein K, Diehl A, Hohwy M, Steuernagel S, Engelke F, Oschkinat H, Reif B (2003) 1H detection in MAS solid state NMR spectroscopy employing pulsed field gradients for residual solvent suppression. J Am Chem Soc 125:7788–7789
Chevelkov V, Faelber K, Diehl A, Heinemann U, Oschkinat H, Reif B (2005) Detection of dynamic water molecules in a microcrystalline sample of the SH3 domain of alpha-spectrin by MAS solid-state NMR. J Biomol NMR 31:295–310
Chevelkov V, Rehbein K, Diehl A, Reif B (2006) Ultra-high resolution in proton solid-state NMR at high levels of deuteration. Angew Chem Int Ed 45:3878–3881
Chevelkov V, Diehl A, Reif B (2007a) Quantitative measurement of differential 15N-Hα/β T2 relaxation times in a perdeuterated protein by MAS solid-state NMR spectroscopy. Magn Res Chem 45:S156–S160
Chevelkov V, Faelber K, Schrey A, Rehbein K, Diehl A, Reif B (2007b) Differential line broadening in MAS solid-state NMR due to dynamic interference. J Am Chem Soc 129:10195–10200
Chevelkov V, Zhuravleva AV, Xue Y, Reif B, Skrynnikov NR (2007c) Combined analysis of 15N relaxation data from solid- and solution-state NMR spectroscopy. J Am Chem Soc 129:12594–12595
Chevelkov V, Diehl A, Reif B (2008) Measurement of 15N–T1 relaxation rates in a perdeuterated protein by MAS solid-state NMR spectroscopy. J Chem Phys 128:052316
Chevelkov V, Fink U, Reif B (2009) Accurate determination of order parameters from 1H,15N dipolar couplings in MAS solid-state NMR experiments. J Am Chem Soc (submitted)
Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM (1990) Deviations from the Simple 2-parameter model-free approach to the interpretation of N-15 nuclear magnetic relaxation of proteins. J Am Chem Soc 112:4989–4991
Cole HBR, Torchia DA (1991) An NMR-study of the backbone dynamics of Staphylococcal nuclease in the crystalline state. Chem Phys 158:271–281
Dvinskikh SV, Zimmermann H, Maliniak A, Sandstrom D (2003) Heteronuclear dipolar recoupling in liquid crystals and solids by PISEMA-type pulse sequences. J Magn Reson 164:165–170
Dvinskikh SV, Zimmermann H, Maliniak A, Sandström D (2005) Heteronuclear dipolar recoupling in solid-state nuclear magnetic resonance by amplitude-, phase-, and frequency-modulated Lee–Goldburg cross-polarization. J Chem Phys 122:044512
Eichmüller C, Skrynnikov NR (2005) A new amide proton R-1 rho experiment permits accurate characterization of microsecond time-scale conformational exchange. J Biomol NMR 32:281–293
Eichmüller C, Skrynnikov NR (2007) Observation of mu s time-scale protein dynamics in the presence of Ln(3+)stop ions: application to the N-terminal domain of cardiac troponin C. J Biomol NMR 37:79–95
Eisenmesser EZ, Bosco DA, Akke M, Kern D (2002) Enzyme dynamics during catalysis. Science 295:1520–1523
Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Gurmukh S, Rienstra CM (2005) Magic-angle spinning solid-state NMR spectroscopy of the beta 1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J Am Chem Soc 127:12291–12305
Frederick KK, Marlow MS, Valentine KG, Wand AJ (2007) Conformational entropy in molecular recognition by proteins. Nature 448:325–330
Fushman D, Cowburn D (1998) Model-independent analysis of 15N chemical shift anisotropy from NMR relaxation data. Ubiquitin as a test example. J Am Chem Soc 120:7109–7110
Giraud N, Böckmann A, Lesage A, Penin F, Blackledge M, Emsley L (2004) Site-specific backbone dynamics from a crystalline protein by solid-state NMR spectroscopy. J Am Chem Soc 126:11422–11423
Giraud N, Blackledge M, Goldman M, Böckmann A, Lesage A, Penin F, Emsley L (2005) Quantitative analysis of backbone dynamics in a crystalline protein from nitrogen-15 spin-lattice relaxation. J Am Chem Soc 127:18190–18201
Giraud N, Sein J, Pintacuda G, Böckmann A, Lesage A, Blackledge M, Emsley L (2006) Observation of heteronuclear overhauser effects confirms the 15N–1H dipolar relaxation mechanism in a crystalline protein. J Am Chem Soc 128:12398–12399
Giraud N, Blackledge M, Böckmann A, Emsley L (2007) The influence of nitrogen-15 proton-driven spin diffusion on the measurement of nitrogen-15 longitudinal relaxation times. J Magn Reson 184:51–61
Grey MJ, Tang YF, Alexov E, McKnight CJ, Raleigh DP, Palmer AG (2006) Characterizing a partially folded intermediate of the villin headpiece domain under non-denaturing conditions: contribution of His41 to the pH-dependent stability of the N-terminal subdomain. J Mol Biol 355:1078–1094
Hall JB, Fushman D (2006) Variability of the N-15 chemical shielding tensors in the B3 domain of protein G from N-15 relaxation measurements at several fields. Implications for backbone order parameters. J Am Chem Soc 128:7855–7870
Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP (2008) Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci USA 105:6284–6289
Henzler-Wildman K, Kern D (2007) Dynamic personalities of proteins. Nature 450:964–972
Hologne M, Faelber K, Diehl A, Reif B (2005) Characterization of dynamics of perdeuterated proteins by MAS solid-state NMR. J Am Chem Soc 127:11208–11209
Hologne M, Chen Z, Reif B (2006a) Characterization of dynamic processes using deuterium in uniformly 2H, 13C, 15N enriched peptides by MAS solid-state NMR. J Magn Res 179:20–28
Hologne M, Chevelkov V, Reif B (2006b) Deuteration of peptides and proteins in MAS solid-state NMR. Prog NMR Spect 48:211–232
Hoogstraten CG, Wank JR, Pardi A (2000) Active site dynamics in the lead-dependent ribozyme. Biochemistry 39:9951–9958
Huster D, Xiao LS, Hong M (2001) Solid-state NMR investigation of the dynamics of the soluble and membrane-bound colicin Ia channel-forming domain. Biochemistry 40:7662–7674
Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE (2004) Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature 430:586–590
Lakomek NA, Fares C, Becker S, Carlomagno T, Meiler J, Griesinger C (2005) Side-chain orientation and hydrogen-bonding imprint supra-tauc motion on the protein backbone of ubiquitin. Angew Chem Int Ed 117:7954–7956
Lange OF, Lakomek N-A, Fares C, Schroeder GF, Walter KFA, Becker S, Meiler J, Grubmueller H, Griesinger C, de Groot BL (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320:1471–1475
Linser R, Fink U, Reif B (2008) Proton-detected scalar coupling based assignment strategies in MAS solid-state NMR spectroscopy applied to perdeuterated proteins. J Magn Reson 193:89–93
Lipari G, Szabo A (1981) Pade approximants to correlation-functions for restricted rotational diffusion. J Chem Phys 75:2971–2976
Lipari G, Szabo A (1982) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc 104:4546–4559
Lorieau JL, McDermott AE (2006) Conformational flexibility of a microcrystalline globular protein: order parameters by solid-state NMR spectroscopy. J Am Chem Soc 128:11505–11512
Lorieau JL, Day LA, McDermott AE (2008) Conformational dynamics of an intact virus: order parameters for the coat protein of Pf1 bacteriophage. Proc Natl Acad Sci USA 105:10366–10371
Mack JW, Usha MG, Long J, Griffin RG, Wittebort RJ (2000) Backbone motions in a crystalline protein from field-dependent H-2-NMR relaxation and line-shape analysis. Biopolymers 53:9–18
Maricq MM, Waugh JS (1979) NMR in rotating solids. J Chem Phys 70:3300–3316
Martinez JC, Pisabarro T, Serrano L (1998) Obligatory steps in protein folding and the conformational diversity of the transition state. Nat Struct Biol 5:721–729
Palmer-III AG, Williams J, McDermott AE (1996) Nuclear magnetic resonance studies of biopolymer dynamics. J Phys Chem 100:13293–13310
Pauli J, Van Rossum B-J, Förster H, De Groot HJM, Oschkinat H (2000) Sample optimization and identification of signal patterns of amino acid side chains in 2D-RFDR spectra of the a-spectrin SH3 domain. J Magn Reson 143:411–416
Redfield AG (1957) On the theory of relaxation processes. IBM J Res Dev 1:19–31
Reif B, Griffin RG (2003) 1H detected 1H, 15N correlation spectroscopy in rotating solids. J Magn Reson 160:78–83
Reif B, Jaroniec CP, Rienstra CM, Hohwy M, Griffin RG (2001) 1H–1H MAS correlation spectroscopy and distance measurements in a deuterated peptide. J Magn Reson 151:320–327
Reif B, Xue Y, Agarwal V, Pavlova MS, Hologne M, Diehl A, Ryabov YE, Skrynnikov NR (2006) Protein side-chain dynamics observed by solution- and solid-state NMR: comparative analysis of methyl 2H relaxation data. J Am Chem Soc 128:12354–12355
Sackewitz M, Scheidt HA, Lodderstedt G, Schierhorn A, Schwarz E, Huster D (2008) Structural and dynamical characterization of fibrils from a disease-associated alanine expansion domain using proteolysis and solid-state NMR spectroscopy. J Am Chem Soc 130:7172–7173
Skrynnikov NR (2007) Asymmetric doublets in MAS NMR: coherent and incoherent mechanisms. Magn Res Chem 45:S161–S173
Tjandra N, Szabo A, Bax A (1996) Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J Am Chem Soc 118:6986–6991
Torchia DA, Szabo A (1982) Spin-lattice relaxation in solids. J Magn Reson 49:107–121
Vallurupalli P, Hansen DF, Kay LE (2008) Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci USA 105:11766–11771
Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH (2008) Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 319:1523–1526
Williams JC, McDermott AE (1995) Dynamics of the flexible loop of triosephosphate isomerase—the loop motion is not ligand-gated. Biochemistry 34:8309–8319
Wu XL, Zilm KW (1993) Cross-polarization with high-speed magic-angle spinning. J Magn Reson A 104:154–165
Wylie BJ, Franks WT, Rienstra CM (2006) Determinations of N-15 chemical shift anisotropy magnitudes in a uniformly N-15, C-13-labeled microcrystalline protein by three-dimensional magic-angle spinning nuclear magnetic resonance spectroscopy. J Phys Chem B 110:10926–10936
Wylie BJ, Sperling LJ, Frericks HL, Shah GJ, Franks WT, Rienstra CM (2007) Chemical-shift anisotropy measurements of amide and carbonyl resonances in a microcrystalline protein with slow magic-angle spinning NMR spectroscopy. J Am Chem Soc 129:5318–5319
Xu J, Xue Y, Skrynnikov NR (2009) Detection of nanosecond time scale side-chain jumps in water/glycerol solvent. J Biomol NMR (in press) (topical issue on dynamics)
Yao L, Vögeli B, Ying J, Bax A (2008) NMR determination of amide N–H equilibrium bond length from concerted dipolar coupling measurements. J Am Chem Soc 130:16518–16520
Zeeb M, Jacob MH, Schindler T, Balbach J (2003) N-15 relaxation study of the cold shock protein CspB at various solvent viscosities. J Biomol NMR 27:221–234
Zhang Q, Sun XY, Watt ED, Al-Hashimi HM (2006) Resolving the motional modes that code for RNA adaptation. Science 311:653–656
Acknowledgments
We thank Nikolai Skrynnikov for many stimulating discussions. This work was supported by the Leibniz-Gemeinschaft and the DFG (grants Re1435, SFB 449, SFB 740).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chevelkov, V., Fink, U. & Reif, B. Quantitative analysis of backbone motion in proteins using MAS solid-state NMR spectroscopy. J Biomol NMR 45, 197–206 (2009). https://doi.org/10.1007/s10858-009-9348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-009-9348-5