
Abstract
The tetratricopeptide repeat (TPR) is a 34-amino acid helix-turn-helix motif that occurs in tandem arrays in numerous proteins. Here we compare the backbone dynamics of a natural 3-repeat TPR domain, from the protein UBP, with the behavior of a designed protein CTPR3, which consists of three identical consensus TPR units. Although the three tandem TPR repeats in both CTPR3 and UBP behave as a single unit, with no evidence of independent repeat motions, the data indicate that certain positions in UBP are significantly more flexible than are the corresponding positions in CTPR3. Most of the dynamical changes occur at or adjacent to positions that are involved in intra-repeat packing interactions. These observations lead us to suggest that the three-TPR domain of UBP does not incorporate optimized packing, compared to that seen in the idealized CTPR. The natural TPR domain is not only less stable overall than CTPR3, but also presents increased local flexibility at the positions where the sequences differs from the conserved consensus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tetratricopeptide repeat (TPR) is a 34-amino acid helix-turn-helix motif found in a tandem repeat arrangement in numerous proteins involved in a variety of cellular processes (Blatch and Lassle 1999; D’Andrea and Regan 2003). Repeat proteins, including TPRs are a class of proteins in which a structural motif, consisting of small numbers of secondary structure elements packed directly against each other, is present in multiple copies in tandem. We have previously described the design and characterization of a series of proteins in which multiple copies of a consensus TPR motif are repeated in tandem. These proteins are named CTPRn, for Consensus TPR, where n is the number of tandem CTPR repeats. All the CTPR proteins undergo cooperative unfolding transitions that can be described by an Ising model in which each repeat has the same intrinsic stability and each pair of adjacent helices has the same favorable energy of interaction (Kajander et al. 2005). Crystal structures indicate that the proteins CTPR2 and CTPR3 have well-defined folded structures (Main et al. 2003) and our previous analysis of NMR relaxation data indicates that the backbones of these proteins have similar rigidity to typical globular proteins (Cheng et al. 2006). Here we compare the behavior of CTPR3 with that of a natural three-TPR domain.
Vpu-binding protein (UBP) is a human cellular protein which has been proposed to interact with a number of other proteins, including HIV-1 viral protein U (Vpu) and HIV-1 Gag; the nonstructural protein NS1 of parvovirus H-1 and cellular chaperones Hsc70 and Hsp90 (Callahan et al. 1998; Cziepluch et al. 1998; Liu et al. 1999; Liou and Wang 2005). The 3-TPR domain of UBP is less thermally stable than CTPR3; their melting temperatures are 59 and 83°C, respectively.
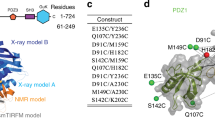
Herein we report a comparison of the backbone motions of the 3-TPR domain of UBP with those of CTPR3, measured by 15N relaxation. Figure 1 shows the structure of CTPR3 (Fig. 1a), an alignment of the sequences of CTPR3 and UBP, the amino acid numbering for both proteins (see legend for explanation of numbering), and the nomenclature used to refer to the α-helices (Fig. 1c).

(a) Ribbon representation of the crystal structure of CTPR3 (PDB entry: 1NA0). The first and second helix of each repeat are colored in orange and blue, respectively, whereas the C-terminal solvation helix is colored in green. (b) Overlayed structures of CTPR3 (PDB entry: 1NA0; Main et al. 2003; cyan) and UBP TPR domain (homology model; magenta). (c) Alignment of sequences of CTPR3 and UBP. A schematic above the sequences indicates the amino acid positions in a TPR repeat and nomenclature of the helices. CTPR3 and UBP amino acid numbering are shown on the sides of the sequence of each repeat. The solid lines connect pairs of conserved hydrophobic residues that define the intra-repeat packing interactions. The positions that correspond to the TPR consensus sequence are highlighted in green, the positions at which the consensus sequence is not conserved in UBP are highlighted in yellow and the positions at which the consensus amino acid is present but the packing partner is not conserved are highlighted in orange. The schematic also shows a conserved charge–charge interaction (dotted line) between residues 26 (positive, highlighted in blue) and residues 29 (negative, highlighted in red). (d) Ribbon Structure of one repeat of CTPR3 (PDB entry: 1NA0) depicting in space filling representation the positions of three pairs of interacting residues that exhibit a significant change in order parameters between UBP and CTPR3. Each pair is colored with two different shades of the same color: purple, positions 26 (K in CTPR3, R in UBP) and 29 (E in CTPR3, C in UBP); green, positions 8 (G in CTPR3, A in UBP) and 24 (Y in CTPR3, C in UBP); and orange, positions 11 (Y in CTPR3, Q in UBP) and 20 (A in both CTPR3 and UBP). Note that the structure on the right is rotated 180° around the vertical axis relative to the structure on the left
Materials and methods
Sample preparation
Uniformly 15N-labeled 3-TPR domain of UBP was expressed and purified as previously described (Cortajarena and Regan 2006) and identity was confirmed by mass spectrometry.
NMR measurements
NMR spectra were acquired from a sample consisting of ~1 mM uniformly 15N-labeled UBP in 50 mM K2HPO4, 100 mM NaCl, 10 mM NaN3, 0.1 mM EDTA, H2O:D2O (95:5%), pH 6.5 at 25°C on a Varian Unity INOVA 500 MHz spectrometer equipped with three radiofrequency channels, pulsed field gradients, and a 1H-detect triple resonance (HCN) probe. Backbone amide resonance assignments were made primarily on the basis of published resonance assignments (Pai et al. 2003). However, ambiguities in assignments were resolved using 3D TOCSY-HSQC and NOESY-HSQC spectra, in which the 1H carrier was placed on the water resonance, the 15N carrier was placed at 120 ppm, and the spectral widths were 7,000, 5,300 and 1,350 Hz in the indirect 1H, direct 1HN, and 15N dimensions, respectively (Zhang et al. 1994). The 15N longitudinal relaxation rate (R 1), 15N transverse relaxation rate (R 2), {1H}–15N steady-state nuclear Overhauser enhancement (NOE), and 15N transverse cross-relaxation rate constants (ηxy) were measured using published 2D 1H–15N HSQC-style pulse sequences (Kay et al. 1989; Farrow et al. 1994; Kroenke et al. 1998). The 1H carrier was placed on the water resonance and the 15N carrier was placed at 120 ppm. All experiments were acquired with spectral widths of 7,000 Hz in the 1H dimension and 1,350 Hz in the 15N dimension. Other experimental parameters were the same as those used in a previous study (Seewald et al. 2000), except that NOE spectra were recorded in duplicate rather than triplicate. In particular, relaxation delays used were τ = 11.1*, 55.5, 133.2, 233.1*, 377.4, 555.0*, 888.0 and 1,998.0* ms for the R 1 experiment and 16.8*, 33.5, 67.1*, 100.6, 150.9, 201.2*, 285.1 and 385.7* ms for the R 2 experiment; asterisks indicated duplicated points.
Essentially the same data collection strategy was used for CTPR3 (Cheng et al. 2006).
Analysis of relaxation data
NMR data were processed using FELIX98 (Molecular Simulation, Inc.). All data analysis methods were as previously described (Seewald et al. 2000) unless noted. Backbone amide resonances with partial or complete spectral overlap, weak peak intensity, or ambiguous assignments were excluded from data analyses. Additionally, amide groups with possible conformational exchange on a slow timescale (R 2/η xy ratios exceeding the average by one standard deviation) were excluded from estimation of the rotational diffusion tensor (Fushman and Cowburn 1998); these residues were Thr-96, Ala-125, Asn-130, His-175, Lys-184, Thr-194, Ser-197, Glu-203, and Lys-205. The R 2/R 1 ratios of the remaining 56 of 121 backbone amide groups and the crystal structure coordinates for CTPR3 (PDB file 1NA0) (Main et al. 2003) were used to determine the overall correlation time and rotational diffusion tensor for isotropic, axially symmetric, and fully anisotropic models using the program quadric diffusion (A. G. Palmer III, Columbia University, New York, NY) (Bruschweiler et al. 1995). The most appropriate diffusion tensor was selected using F-statistical analysis and from a comparison of χ2 goodness-of-fit parameters (see "Results" section). The entire TPR domain of UBP was best described by an oblate axially symmetric rotational diffusion tensor. Using this rotational diffusion tensor, initial model-free calculations were executed using the program Modelfree (A. G. Palmer III) which fits relaxation data to each of the five versions of the Lipari-Szabo model-free formalism (Lipari and Szabo 1982a, b; Clore et al. 1990a, b; Barbato et al. 1992) commonly referred to as “models” 1–5. The fitted dynamics parameters for each model are: model 1, order parameter (S 2); model 2, S 2 and internal correlation time (τe); model 3, S 2 and exchange broadening contribution to transverse relaxation (R ex); model 4, S 2, τe, and R ex; model 5, order parameters for two time scales (S f 2 and S 2s ) and τe for the slower time scale. Selection of the model best describing the internal dynamics of each backbone amide group was made using ModelFree as previously described (Mandel et al. 1995). Following model-selection, a final model-free calculation was executed in which all dynamics parameters and the rotational diffusion tensor were simultaneously optimized. Uncertainties in the dynamics parameters were obtained from 500 Monte Carlo simulations carried out by ModelFree.
Homology modeling of the UBP TPR domain
The model of the UBP TPR domain structure was generated using Swiss-model by automated homology modeling (http://swissmodel.expasy.org//SWISS-MODEL.html) (Schwede et al. 2003) including energy minimization. The model is based on the coordinates of the TPR domain of PP5 (PDB ID: 1A17) (Das et al. 1998), CTPR3 (PDB ID: 1NA0) (Main et al. 2003), and the TPR1 domain of Hop (PDB ID: 1ELW) (Scheufler et al. 2000). The output model from Swiss-model server was subjected to 3 × 100 steps of steepest descent energy minimization by using GROMOS 43B1 force field program included in Swiss-pdb Viewer. The quality of the model was checked using MOLPROBITY (http://molprobity.biochem.duke.edu/) (Davis et al. 2007). The model was further validated by comparison of the backbone ψ angles to those determined from the reported Hα, CO, Cα, Cβ and NH chemical shift values (Pai et al. 2003) using the program Talos (Cornilescu et al. 1999).
Results
Sequence and structure conservation in TPR domains: homology model of the UBP TPR domain
TPR domains present highly conserved “TPR signature residues”, that are the key determinants of the TPR fold. The intra-repeat packing is mediated by these residues, with pairs of small and large hydrophobic residues making specific interactions (positions 4 and 27, positions 8 and 24, positions 11 and 20 and positions 15 and 17) (Fig. 1c and Table 1).
Sequence alignments in Clustal W (http://www.ebi.ac.uk/Tools/clustalw2/index.html) (Larkin et al. 2007) show that the UBP TPR domain has 27% amino acid sequence identity with CTPR3, 34% with TPR1 domain of Hop, and 28% with PP5 TPR domain. This high degree of sequence identity and the presence of most of the conserved “signature residues” in the UBP sequence, indicates that the UBP TPR domain will have a structure very similar to the TPR domains used in the homology model generation.
Alignment of the structures of TPR domains that contain three tandem TPR repeats, including CTPR3, the TPR1 domain of Hop and the TPR domain PP5, gives pair-wise backbone RMSD values that vary from 1.1 to 1.9 Å (Cortajarena and Regan 2006). This result emphasizes that the structure of TPR domains is highly conserved. It is important to note that the sequence identity between pairs of these domains, is similar and typically slightly lower, than that of UBP and the structures used for homology modeling (see "Materials and methods" and below). TPR1 domain of Hop has 23% sequence identity with CTPR3 and 27% with the TPR domain of PP5; CTPR3 has 22% sequence identity with the TPR domain of PP5.
We constructed a structural model of the UBP TPR domain based on sequence alignment with the TPR domains of the proteins PP5 (Das et al. 1998), CTPR3 (Main et al. 2003), and the TPR1 domain of Hop (Scheufler et al. 2000) to provide a context in which to interpret dynamical differences between UBP and CTPR3. The final model of the TPR domain of UBP displays good geometry and stereochemical properties, according to the results of the model validation by MOLPROBITY (Davis et al. 2007) (see “Electronic supplementary material”). The backbone ψ angles of the model are in excellent agreement with those determined from the Hα, CO, Cα, Cβ and NH chemical shifts of UBP (Pai et al. 2003) (see “Electronic supplementary material”). The overlay of the UBP model with the structure of CTPR3 gives a backbone RMSD of 1.7 Å (Fig. 1b), with the conserved TPR signature residues located in identical positions in both structures (Fig. 1c). Moreover, in previous studies we showed that the TPR domain of UBP binds the same peptide ligand as the TPR1 domain of HOP. In these studies, we used NMR chemical shift mapping to identify the UBP residues involved in ligand binding. When these residues are mapped onto the model of the UBP TPR structure, they map to the typical concave binding pocket of the TPR (Cortajarena and Regan 2006). This is exactly where the same binding residues are located in the crystal structures of the TPR1 domain of HOP.
Together, these results fully support the validity of the model for the TPR domain of UBP and its use in the interpretation of the relaxation data.
Relaxation parameters
Relaxation parameters were determined for 64 of 121 backbone amide groups in the TPR domain of UBP. The sequence coverage (53%) is similar to that obtained for CTPR3 (48%). Nevertheless, the residues for which data were obtained are well dispersed throughout the protein sequence. TPR motifs 1, 2 and 3 were represented by relaxation data obtained from 19, 18 and 17 residues, respectively, and the carboxy-terminal solvating helix was represented by relaxation data from 10 residues. The R 1, R 2, NOE, R 2/R 1, ηxy and R 2/ηxy values are plotted in Fig. 2a–f and listed in electronic supplementary material.

Relaxation parameters for the TPR domain of UBP as a function of residue number; (a) R 1, (b) R 2, (c) NOE, (d) R 2/R 1, (e) ηxy, and (F) R 2/ηxy (●). The positions of the α-helices are indicated schematically at the top of the figure by solid rectangles
Rotational diffusion of the TPR domain and of individual repeats
Our previous study of CTPR2 and CTPR3 showed that all the TPR repeats in each protein tumble in solution as a single folding unit, rather than each repeat displaying segmental rotational motion (Cheng et al. 2006). This behavior is consistent with the extensive intra- and inter-repeat helix-helix interactions that are observed in the crystal structures of the CTPR proteins (Main et al. 2003; Kajander et al. 2005) and in the structures of natural 3-TPR domains (Das et al. 1998; Scheufler et al. 2000). Strong coupling between repeats is further evidenced by the co-operative chemical and thermal denaturation transitions observed for CTPR2 and CTPR3 (Main et al. 2003). The 3-TPR domain of UBP also displays a single cooperative unfolding transition (Cortajarena and Regan 2006). However, the stability of the UBP domain is considerably lower than that of CTPR3 (Tm of 59 vs. 83°C and ΔGUNF 6.1 vs. 10.7 kcal mol−1) (Main et al. 2003, 2005; Cortajarena and Regan 2006). It was therefore of interest to compare backbone dynamics of CTPR3 and UBP, proteins which both contain three tandem TPR repeats, but which differ significantly in stability.
We used the backbone relaxation data to determine the rotational diffusion of the entire TPR domain of UBP as well as each of the three individual TPR repeats. The data for the entire TPR domain were best represented by an oblate axially symmetric diffusion tensor with an effective rotational correlation time, τm = (2D|| + 4D⊥)−1, of 14.46 ± 0.28 ns (Fexp = 6.16 vs. F0.95 = 2.76 for comparison with an isotropic diffusion tensor; Fexp = 0.12 vs. F0.95 = 3.15 for comparison with a fully anisotropic diffusion tensor). In comparison, data for each individual TPR repeat were best represented by isotropic diffusion tensors (Fexp < F0.95 for comparison with the axially symmetric model). The overall correlation times (τm) for repeats 1 (residues 91–124), 2 (residues 125–158) and 3 (residues 159–192) are 14.79 ± 0.13, 14.26 ± 0.17, and 14.74 ± 0.17 ns, respectively. The similarity of the rotational correlation times of the individual TPR motifs with each other and with that of the entire domain suggests that the motions of the repeats are correlated and that the entire TPR domain of UBP behaves as a single, rigid domain. This conclusion is consistent with the single, cooperative folding transition observed for the TPR domain of UBP (Cortajarena and Regan 2006). It is also the behavior we observed for CTPR2 and CTPR3.
Dynamics parameters
The dynamics of backbone amide bond vectors in the TPR domain of UBP were determined by fitting relaxation data to the Lipari–Szabo model-free formalism (Lipari and Szabo 1982a, b; Clore et al. 1990a, b; Barbato et al. 1992). These calculations yield, for each NH group, the generalized order parameter S 2, which is a measure of the degree of spatial restriction of the bond vector’s motion. The order parameter can range in value from zero to one with an S 2 value of 0 indicating completely unrestricted motions and S 2 value of 1 indicating completely restricted motions. Model-free analysis was performed for the entire TPR domain of UBP using an axially symmetric oblate diffusion tensor, as determined above. Additionally, model-free analyses were performed independently for TPR motifs 1, 2 and 3 using the isotropic diffusion tensor determined separately for each individual repeat. The order parameters determined from the model-free calculations performed on individual repeats did not differ significantly from those determined from calculations performed on the entire TPR domain (data not shown). Thus, from this point on we will only discuss S 2 values obtained from entire domain calculations. As an added control, model-free analyses were performed for the entire TPR domain of UBP using the rotational diffusion tensor previously determined for CTPR3 (Cheng et al. 2006). Order parameters determined using the axial prolate rotational diffusion parameters of CTPR3 were not significantly different from those determined using UBP’s own diffusion tensor (data not shown), thus allowing us to make a fair comparison between the internal dynamics of the TPR domain of UBP and those of CTPR3.
Of the 64 backbone amide groups included in the model-free analysis of the TPR domain of UBP, relaxation data for the majority (62 amide bond vectors) were fit adequately to the simplified version of the model-free formalism (S 2 only), indicating that internal motions affecting 15N relaxation are on a time scale faster than ~20 ps. Relaxation data for two amide bond vectors (Ala-110 and Lys-205) required incorporation of an effective internal correlation time (τe) to account for slightly slower internal motions (τe = 48 ± 22 and 41 ± 17, respectively). All of the data were satisfied without recourse to models describing conformational exchange on the microsecond to millisecond time scale.
Comparison of fast time scale dynamics of UBP with CTPR3
The order parameters for backbone amide groups in the TPR domain of UBP and CTPR3 are shown in Fig. 3a as a function of each protein’s sequence; the complete listing of the values for UBP is included in the Electronic Supplementary Material. The variations in order parameters across the sequence are similar for the two proteins, but the average order parameter for UBP is slightly lower than that of CTPR3 (see inset of Fig. 3a). The S 2 values are 0.840 ± 0.046 for UBP and 0.878 ± 0.046 for CTPR3 (average of the values of 37 equivalent positions, ±standard deviation); these values are significantly different in a paired z-test (p = 0.0004). The order parameters for each helix of CTPR3 and UBP are compared in Table 2 (statistics are for paired t-tests). The most significant increases in backbone flexibility of UBP relative to CTPR3 are located in the B2 helix (p = 0.01, n = 5), the solvation helix (p = 0.04, n = 8), and the B1 helix (p = 0.07, n = 6). Pairwise comparisons of individual residues identify nine residues whose order parameters differ between the two proteins at the ≥90% confidence level (Table 3). All of these are located in the B1 helix, the A2 helix, or the B1-A2 linker. The significant difference between solvation helices of the two proteins appears to be due to small differences throughout this helix rather than large changes at specific positions (Fig. 3a, b).

(a) A comparison of S 2 values for the TPR domain of UBP (●) and CTPR3 (□) as a function of residue number. Inset is a histogram of S 2 values in UBP (thick line) and CTPR3 (thin line). (b) The difference between CTPR3 and UBP S 2 values as a function of residue number. The positions of the α-helices are indicated schematically at the top of each graph by solid rectangles
The increased flexibility on a fast timescale of several backbone amide groups within UBP compared with those found in CTPR3 can be understood by considering the residue specific deviations of the natural TPR domain of UBP from the consensus sequence of CTPR3 (Fig. 1c, Tables 1 and 3). Of particular relevance is the existence in CTPR3 of various conserved interacting residues, which are a feature of TPR motifs in general, but which are not all present in UBP (Table 3, Fig. 1c). Figure 1d shows the structure of a consensus TPR repeat, highlighting several of these interactions. Table 1 lists the amino acids found at these positions in CTPR3 and UBP, the amino acids most frequently found at these positions in TPR repeats, and the frequencies of occurrence for these amino acid types among TPR repeats. The specific interactions are discussed in detail below.
Most of these conserved interactions determine the hydrophobic intra-repeat packing. For example, at position 24, Tyr is statistically the most favored amino acid in all TPRs (Table 1) and it participates in a conserved interaction with the conserved Gly-8 of the same repeat (Table 1 and Fig. 1d). In all three repeats of CTPR3 and in repeats 1 and 3 of UBP, position 24 is Tyr and position 8 is Gly. In repeat 2 of UBP, however, this conserved Tyr/Gly pair is replaced by a Cys-148/Ala-132 pair (Table 3). Cys is rarely found at this position in TPRs, and would not be able to make the conserved ‘large-small/Tyr-Gly’ interaction. Indeed, the homology model of UBP indicates a substantial reduction in the interaction surface between positions 8 and 24 of repeat 2, compared with the corresponding residues in CTPR3 (green in Fig. 1d). The lack of an optimal interaction between residues 148 and 132 provides a plausible explanation for the increase in fast timescale flexibility of the residues adjacent to Cys-148 (Asp-147, Glu-149, and Arg-150). Preliminary analysis of data for Cys-148 also indicated a low order parameter, but we excluded Cys-148 itself from the current discussion because the cross peak for Cys-148 is unusually weak.
There is also a significant enhancement of dynamics at TPR position 20 of the B1 helix (UBP residue Ala-110) compared to the corresponding residue (Ala-28) in CTPR3 (ΔS2 = −0.142 ± 0.060, p = 0.02) (Table 3). The Ala at this position participates in a conserved interaction with a large hydrophobic amino acid (most commonly Tyr) at position 11 of the same repeat; these residues are colored orange in Fig. 1d. This interaction is present in all three repeats of CTPR3 and in repeats 1 and 2 of UBP, but in repeat 3 of UBP the Tyr is replaced by Gln. We suggest that the enhanced dynamics observed for position 20 of repeat 1 of UBP are because the favorable interaction of Ala at this position with Tyr at position 11 is not present. The manifestation of changes in the dynamics of the partner residue to Gln11 provides support for the model in which residues 11 and 20 interact. In all the repeats of CTPR3, and in repeats 1 and 2 of UBP there is a favorable Tyr11-Ala20 interaction. Repeat 3 of UBP substitutes the much less favored Gln11-Ala20 interaction, and as a consequence the dynamics of Ala20 are increased.
In addition to the disruption of hydrophobic interactions, UBP also lacks a conserved charge-charge interaction between position 26 and position 29 of repeat 2. Covariation analyses show that when a positively charged residue, such as Arg or Lys, is present at position 26, there is a greater chance of having a negatively charged residue, Asp or Glu at position 29, and vice versa (Magliery and Regan 2004). This type of charge–charge interaction is conserved in CTPR3, where all three repeats have Lys at position 26 and Glu at position 29 (purple in Fig. 1d). Such a Lys-Glu pair is also found at positions 26 and 29 in repeats 1 and 3 of UBP. In repeat 2 of UBP, however, this charge–charge interaction is not present (Table 3). Position 26 is Arg, but position 29 is Cys, which occurs at extremely low frequency at this position in TPRs in general (Table 1). Removal of the favorable electrostatic interaction between residues 26 and 29 in repeat 2 of UBP provides a reasonable explanation for the considerable increase in flexibility of Arg-150 at position 26 (ΔS 2 = −0.148 ± 0.071, p = 0.04) and would be expected to contribute to the increased flexibility of other residues in the same turn of helix B2.
The general observation from the dynamics data is that if one residue of a pair of interacting residues is not conserved the consequence in the flexibility can show up in either the region of the mutated residue (for example in residues adjacent to Cys-148) or in its partner (for example in Ala-110 and Arg-150). This latter effect is additional evidence for the disruption of a packing interaction.
Finally, two residues in the B1-A2 linker (Asn-121 and Ala-125) show minor increases in flexibility in UBP relative to CTPR3 (0.05 < p < 0.1). Interestingly, mutation of the linker from Asp-Pro-Asn-Asn to Asp-Pro-Arg-Ser in the context of CTPR3 causes a significant decrease in stability (Main et al. 2003; Kajander et al. 2005). This result is consistent with the observation of increased flexibility in the B1-A2 linker of UBP (Asn-Pro-Ala-Asn) versus that in CTPR3 (Asp-Pro-Asn-Asn). Detailed comparisons at other positions in this linker and at most positions in the B2-A3 and B3-S linkers are prevented due to resonance overlap.
Discussion
First, our results show that in both CTPR3 and UBP the 3-TPR units are coupled and show no evidence of uncorrelated inter-repeat motion. Second, although CTPR3 has a significantly higher melting temperature than UBP, under conditions of these measurements (25°C) the dynamic behavior of most residues is the same. Third, there are several clearly observable local differences in the dynamics of CTPR3 and UBP. Certain residues in UBP are significantly more flexible than residues at the corresponding positions in CTPR3. Most of these dynamic changes can be explained by considering amino acid differences that influence key intra-repeat packing interactions. The consensus design of the CTPR unit incorporates the most prevalent amino acid at each position in the sequence, and simultaneously introduces conserved inter-residue interactions. The data we present provide evidence for the existence of these interactions and show how they influence the dynamic properties of the protein in solution. We observe specific examples of non-optimal packing in UBP, which are associated with an increase in mobility in the vicinity of the ‘defect’. In CTPR3 all the repeats are identical to each other, all have the same inter-residue packing interactions, and all show the same dynamic behavior. By contrast, the three TPR repeats in UBP are all different, and those which deviate from the consensus sequence and consensus packing interactions, show local changes in dynamics that reflect these differences. The possible functional significance of these differences will be the focus of future studies.
Electronic supplementary material available
Two tables listing the relaxation data and model-free dynamics parameters for the TPR domain of UBP; a table of results obtained from MOLPROBITY for the UBP structural model; the assigned HSQC spectrum for UBP; and a figure comparing the backbone ψ angles of the model with those determined from the UBP chemical shifts.
Abbreviations
- ηxy :
-
Cross-relaxation rate constant
- HSQC:
-
Heteronuclear single quantum coherence
- MSA:
-
Multiple sequence alignment
- NMR:
-
Nuclear magnetic resonance
- NOE:
-
Nuclear Overhauser effect
- R 1 :
-
Longitudinal relaxation rate constant
- R 2 :
-
Transverse relaxation rate constant
- R ex :
-
Conformational exchange rate
- S 2 :
-
Order parameter
- τe :
-
Internal correlation time
- τm :
-
Rotational correlation time
- UBP:
-
Vpu-binding protein
- TPR:
-
Tetratricopeptide repeat
- CTPR:
-
Consensus TPR
References
Barbato G, Ikura M, Kay LE, Pastor RW, Bax A (1992) Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31:5269–5278
Blatch GL, Lassle M (1999) The tetratricopeptide repeat: a structural motif mediating protein–protein interactions. Bioessays 21:932–939
Bruschweiler R, Liao XB, Wright PE (1995) Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268:886–889
Callahan MA, Handley MA, Lee YH, Talbot KJ, Harper JW, Panganiban AT (1998) Functional interaction of human immunodeficiency virus type 1 Vpu and Gag with a novel member of the tetratricopeptide repeat protein family. J Virol 72:5189–5197
Cheng CY, Jarymowycz VA, Cortajarena AL, Regan L, Stone MJ (2006) Domain motions and backbone flexibility in designed proteins with different numbers of identical consensus tetratricopeptide repeats. Biochemistry 45:12173–12183
Clore GM, Driscoll PC, Wingfield PT, Gronenborn AM (1990a) Analysis of the backbone dynamics of interleukin-1 beta using two-dimensional inverse detected heteronuclear 15N–1H NMR spectroscopy. Biochemistry 29:7387–7401
Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM (1990b) Deviations from the simple 2-parameter model-free approach to the interpretation of 15N nuclear magnetic-relaxation of proteins. J Am Chem Soc 112:4989–4991
Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302
Cortajarena AL, Regan L (2006) Ligand binding by TPR domains. Protein Sci 15:1193–1198
Cziepluch C, Kordes E, Poirey R, Grewenig A, Rommelaere J, Jauniaux JC (1998) Identification of a novel cellular TPR-containing protein, SGT, that interacts with the nonstructural protein NS1 of parvovirus H-1. J Virol 72:4149–4156
D’Andrea LD, Regan L (2003) TPR proteins: the versatile helix. Trends Biochem Sci 28:655–662
Das AK, Cohen PW, Barford D (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J 17:1192–1199
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LM, Arendall WBIII, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383
Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33:5984–6003
Fushman D, Cowburn D (1998) Model-independent analysis of 15N chemical shift anisotropy from NMR relaxation data. Ubiquitin as a test example. J Am Chem Soc 120:7109–7110
Kajander T, Cortajarena AL, Main ER, Mochrie SG, Regan L (2005) A new folding paradigm for repeat proteins. J Am Chem Soc 127:10188–10190
Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28:8972–8979
Kroenke CD, Loria JP, Lee LK, Rance M, Palmer AG (1998) Longitudinal and transverse 1H–15N dipolar 15N chemical shift anisotropy relaxation interference: unambiguous determination of rotational diffusion tensors and chemical exchange effects in biological macromolecules. J Am Chem Soc 120:7905–7915
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) ClustalW2 and ClustalX version 2. Bioinformatics 23:2947–2948
Liou ST, Wang C (2005) Small glutamine-rich tetratricopeptide repeat-containing protein is composed of three structural units with distinct functions. Arch Biochem Biophys 435:253–263
Lipari G, Szabo A (1982a) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc 104:4546–4559
Lipari G, Szabo A (1982b) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc 104:4559–4570
Liu FH, Wu SJ, Hu SM, Hsiao CD, Wang C (1999) Specific interaction of the 70-kDa heat shock cognate protein with the tetratricopeptide repeats. J Biol Chem 274:34425–34432
Magliery TJ, Regan L (2004) Beyond consensus: statistical free energies reveal hidden interactions in the design of a TPR motif. J Mol Biol 343:731–745
Main ER, Stott K, Jackson SE, Regan L (2005) Local and long-range stability in tandemly arrayed tetratricopeptide repeats. Proc Natl Acad Sci USA 102:5721–5726
Main ERG, Xiong Y, Cocco MJ, D’Andrea L, Regan L (2003) Design of stable alpha-helical arrays from an idealized TPR motif. Structure 11:497–508
Mandel AM, Akke M, Palmer AGIII (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol 246:144–163
Pai MT, Yang CS, Tzeng SR, Wang C, Cheng JW (2003) 1H, 15N and 13C resonance assignments of the tetratricopeptide repeat (TPR) domain of hSGT. J Biomol NMR 26:381–382
Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210
Schwede T, Kopp J, Guex N, Peitsch M (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucl Acids Res 31:3381–3385
Seewald MJ, Pichumani K, Stowell C, Tibbals BV, Regan L, Stone MJ (2000) The role of backbone conformational heat capacity in protein stability: temperature dependent dynamics of the B1 domain of Streptococcal protein G. Protein Sci 9:1177–1193
Zhang OW, Kay LE, Olivier JP, Forman-Kay JD (1994) Backbone 1H and 15N resonance assignments of the N-terminal Sh3 domain of Drk in folded and unfolded states using enhanced-sensitivity pulsed-field gradient NMR techniques. J Biomol NMR 4:845–858
Acknowledgements
We thank the reviewers and editor for helpful suggestions. We thank Drs. Douglas E. Brown and John W. Tomaszewski (Indiana University NMR Facility) for discussions and Drs. Lewis Kay (University of Toronto) and Mark Rance (University of Cincinnati) for providing pulse programs. This work was supported in part by a grant awarded to M. J. S. from the National Science Foundation (MCB-0212746), and by Human Frontier Science Program grant RGP0044 to LR and also by a sub-contract to LR from NIH grant number GM080515.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Jarymowycz, V.A., Cortajarena, A.L., Regan, L. et al. Comparison of the backbone dynamics of a natural and a consensus designed 3-TPR domain. J Biomol NMR 41, 169–178 (2008). https://doi.org/10.1007/s10858-008-9250-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-008-9250-6