
Abstract
Molecular size has limited solution NMR analyses of proteins. We report 13C–13C NOESY experiments on a 480 kDa protein, the multi-subunit ferritin nanocage with gated pores. By exploiting 13C-resonance-specific chemical shifts and spin diffusion effects, we identified 75% of the amino acids, with intraresidue C–C connectivities between nuclei separated by 1–4 bonds. These results show the potential of 13C–13C NOESY for solution studies of molecular assemblies >100 kDa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The number of solution structures determined by NMR has been growing rapidly since the first NMR protein structure determination in 1985 (Kaptein et al. 1985; Williamson et al. 1985). Complete structure determinations by solution NMR have generally been restricted to proteins of relatively low molecular weight (< 30 kDa) because of transverse relaxation rates and thus signal linewidths, which increase with the size of the macromolecule. For proteins of < 100 kDa (Frueh et al. 2006), novel techniques using cross-correlated relaxation between dipole–dipole interactions and chemical shift anisotropy (CSA) have overcome the limitations imposed by transverse relaxation, allowing the detection of some sets of signals (Fernández and Wider 2003; Frueh et al. 2006; Wider 2005). Examples are transverse relaxation-optimised spectroscopy (TROSY), (Pervushin et al. 1997) cross-correlated relaxation-enhanced polarization transfer (CRINEPT), (Riek et al. 1999) and cross-correlated relaxation-induced polarization transfer (CRIPT) (Brüschweiler and Ernst 1992; Dalvit 1992; Riek et al. 1999). TROSY-experiments, in combination with selective reprotonation schemes of perdeuterated proteins, have been used to assign 13C and 1H chemical shifts from methyl (Tugarinov et al. 2003) and methylene (Miclet et al. 2004; Tugarinov et al. 2005b) groups in proteins below 100 kDa. 3D and 4D TROSY-based NOESY spectra have been used to measure HN–methyl and methyl–methyl distances in a 82 kDa protein, allowing the definition of its global fold in solution (Tugarinov et al. 2005a). CRINEPT- and CRIPT-based 1H-15N correlation experiments, in combination with extensive protein deuteration, have successfully been used for the detection of amide resonances in systems up to 900 kDa (Flaux et al. 2002). Amide resonances are extremely useful for monitoring conformational changes and intermolecular interactions in proteins. Given the lack of sequential and residue-type information for systems larger than 100 kDa, the use of 1H–15N correlation experiments for studying intermolecular interactions in supramolecular assemblies has been limited to those cases where the assignment for the lower molecular weight component is available and the shift changes can be monitored upon formation of the complex. For this type of complexes, the use of [1H–1H]-NOESY-[15N–1H]-CRINEPT-HMQC for the detection of cross peaks between backbone amides has been proposed very recently (Horst et al. 2006). They provide precious structural information.
An alternative and complementary approach to 1H-detected NMR, is represented by 13C-direct detection experiments (Bermel et al. 2003; Bermel et al. 2005; Bermel et al. 2006b; Bermel et al. 2006a; Bertini et al. 2004b; Bertini et al. 2004c; Bertini et al. 2004a). In these cases the smaller magnetic moment of 13C with respect to 1H reflects in a reduction of signal linewidth. 13C-13C NOESY-type experiments (Bertini et al. 2004b; Bertini et al. 2004c) are the most likely to alleviate the negative effect of the molecular size, even more than 1H-detected 13C–13C magnetization transfer (Fischer et al. 1996). Indeed, the coherence transfer at the basis of experiments relying on scalar couplings suffers for the reduced transverse relaxation times in high molecular weight systems. On the contrary, the magnetization transfer phenomena at the basis of NOESY experiments occur when the magnetization is along the z-axis, and are therefore not affected by transverse relaxation (Banci et al. 1991; Bertini et al. 2004c). During the NOESY mixing time two processes are operative: the cross-relaxation, that is responsible of the magnetization transfer through dipolar coupling, and the longitudinal relaxation which restores the magnetization to the equilibrium values. The longitudinal relaxation times are substantially longer in large proteins. The cross relaxation increases with molecular weight, being directly proportional to the rotational correlation time of the molecule (Neuhaus and Williamson 1989). Therefore the NOESY intensities gain from both processes. The advantage of using 13C–13C NOESY experiments for systems of higher molecular weight was proposed some years ago and illustrated by the comparison of the spectra for monomeric (16 kDa) and dimeric (32 kDa) forms of the protein superoxide dismutase (SOD) (Bertini et al. 2004b; Bertini et al. 2004c). In the dimeric protein, all the expected Cα–CO connectivities were detected with higher intensity than in the monomeric protein. In addition, most of the two-bond CO–Cβ cross peaks were observed for the dimeric SOD, when long mixing times were used (Bertini et al. 2004b). Therefore, we decided to test the use of 13C-13C NOESY for the detection of one and two-bond carbon correlations in a large protein (>100 kDa), the ferritin nanocage with gated pores (480 kDa).
Materials and methods
Ferritin was expressed in BL21 DE3 pLys E. coli cells in 2H, 13C,15N- labelled Spectra9 medium from Spectra Stable Isotopes (Fiaux et al. 2004). The protein was treated for iron removal. The purified protein was 90% deuterated, based on MALDI experiments.
The correlation tumbling time of the protein in solution at the concentration of the NMR sample was checked by NMRD (nuclear magnetic relaxation dispersion) measurements and found to be 170 ns at 298 K, as expected for a spherical molecule of this size (Bertini et al. 2000).
The 13C–13C NOESY experiments were carried out on a 16.4 T Bruker AVANCE 700 spectrometer equipped with a triple-resonance TXO probe optimised for 13C direct-detection experiments, at 298 K on a sample that was 2 mM in monomer concentration, at pH 7.5 (20 mM phosphate buffer). Composite pulse decoupling on 1H and 2H was applied during the whole duration of the experiments (Bertini et al. 2004b). 13C–13C NOESY maps were recorded at four different mixing times (150 ms, 300 ms, 500 ms, 1 s) on the full spectral width and on the aliphatic region in order to increase resolution. Experiments on a sample, 2 mM in monomer, lasted 40 h each to achieve good signal-to-noise ratios.
Results and discussion
The ferritin M protein from bullfrog was chosen as a test case. The protein is a homo polymer of 24 subunits (Trikha et al. 1995). Each subunit has 176 amino acids folded into four helix bundles; in living cells the cavity contains concentrated iron (hydrated ferric oxide mineral). The protein is assembled as a spherical cage with an external diameter of 12 nm, and an inner cavity, 8 nm in diameter (Fig. 1) (Ha et al. 1999; Liu and Theil 2005). The overall molecular weight is 480 kDa.

Ribbon representation of the cross-section of the ferritin protein nanocage structure, viewed down the three-fold axis, and of the monomeric unit (PDB ID 1MFR)
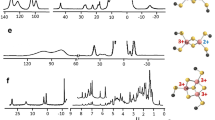
In 13C–13C NOESY maps intraresidue connectivities could be easily detected for most residues (Fig. 2). At shorter mixing times the most intense peaks are those between carbon nuclei connected by a single bond. Connectivities between methionine Cɛ and Cγ (which correspond to nucleus–nucleus distances of about 2.5 Å), and between arginine Cζ and Cδ, (which correspond to nucleus–nucleus distances of about 2.7 Å), were also observed, although they were extremely weak. For connectivities among carbon nuclei separated by two, three, or four C–C bonds, we took advantage of the spin-diffusion effects which increase in intensity with longer mixing times. It appears that spin diffusion is driven by bound 13C nuclei (Fig. 3), rather than through 13C nuclei belonging to different amino acids, due to the relatively large distances among the latter (see below). For example in the case of Ile, cross peaks between all the possible cross peaks connecting pairs of carbon nuclei different from CO, were detected (Fig. 3). The maximum intensity for connectivities between carbons bound directly to each other occurs at 300 ms mixing time, whereas for those between carbons separated by two bonds it occurs at 500 ms (Fig. 3). By taking advantage of the residue-specific chemical shifts of carbon nuclei resonances, we have been able to clearly detect the spin-patterns of most aliphatic residues, using intraresidue patterns of NOESY connectivities. Some aromatic residues (7 His and 1 Tyr) could be identified using Cβ–Cγ cross peaks. In addition, the Cβ–Cδ2 cross peaks of histidine were also detected (Fig. 4). This first inspection allowed the easy identification of a number of spin patterns, accounting for 75% of total amino acids (as summarized in Table 1). The chemical shift index analysis based on Cα and Cβ, permitted the identification of the secondary structure element to which each amino acid belongs (Table 1). Good agreement between CSI analysis and the X-ray crystal structure of ferritin supported the accuracy of the assignments and indicated the detection of amino acids located throughout the entire protein structure. While cross peaks between aliphatic signals (Fig. 2) and between aliphatic and aromatic signals (Fig. 4) were often well resolved, the carbonyl signals were extremely broad. This effect, due to chemical shift anisotropy relaxation, gives rise to unresolved cross peaks and prevents identification of backbone connectivities. The shortest inter-residue C–C distances in ferritin (and in all proteins in general), are those between carbonyls and carbonyls and Cα of sequential amino acids. Such distances are as short as 2.4–2.9 Å. At the current stage of spectral resolution achieved we could not determine with certainty the presence of CO–CO peaks, given their large linewidths. The CO–Cα region was difficult to analyze for the same reason. In the X-ray crystal structure of ferritin, all the other interresidue connectivities involving pairs of carbon atoms are longer that 3.2 Å. None of these could be observed under the used experimental conditions.

Aliphatic region of the 13C–13C NOESY spectrum recorded with 500 ms mixing time. Several peaks are color-coded to exemplify residue-type assignment

Examples of the NOESY pattern for Ile, Val, and Ala residues and corresponding build-up curves for selected connectivities

Portion of the 13C–13C NOESY map containing Cβ–Cγ and Cβ–Cδ2 cross peaks for His residues
Conclusions
The present results show that spin diffusion in a large protein such as ferritin is confined to coupled spins of the same residue and represents a strong advantage for 13C–13C NOESY experiments. In a large protein such as ferritin, the 13C–13C NOESY solution spectra are rich in information, as emphasized by the large number of resolved peaks, even for an alpha-helical protein as ferritin. Per residue assignment is possible for those amino acids that possess peculiar 13C chemical shits. Identification, by 13C–13C NOESY, of the carbon nuclei in a given type of amino acid of a large protein is similar to the information obtainable from TOCSY experiments of low molecular weight proteins. There are also several advantages of 13C–13C-NOESY experiments compared to those limited to amide resonances, such as elimination of problems related to amide proton exchange with bulk solvent and the ability to monitor the full side chain of the amino acids. The ability to monitor side chains, to assign them to a given amino-acid and secondary structure-type element, represents a step forward from simply monitoring backbone amides and extends to high molecular weight systems the potential of solution NMR to characterize intermolecular interactions and conformational changes.
Future investigations outside the scope of this study aim at improved spectral resolution, for more extensive and possibly sequence-specific assignment, as well as at increased sensitivity of the experiments, to facilitate detection of long-range interresidue connectivities. However, even at the present stage of development, the 13C–13C NOESY methodology can be of great help for spectral analyses of many high molecular weight proteins or supramolecular adducts and contributes to lower the macromolecular size barrier for solution NMR. The novel per residue assignment here described facilitates studies of large molecular assemblies and provides a complementary/alternative approach to the 1H-based NMR spectroscopy.
References
Banci L, Bertini I, Luchinat C (1991) Nuclear and electron relaxation. The magnetic nucleus-unpaired electron coupling in solution. VCH, Weinheim
Bermel W, Bertini I, Duma L, Emsley L, Felli IC, Pierattelli R, Vasos PR (2005) Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angew Chem Int Ed 44:3089–3092
Bermel W, Bertini I, Felli IC, Kümmerle R, Pierattelli R (2003) 13C direct detection experiments on the paramagnetic oxidized monomeric copper, zinc superoxide dismutase. J Am Chem Soc 125:16423–16429
Bermel W, Bertini I, Felli IC, Lee Y-M, Luchinat C, Pierattelli R (2006a) Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J Am Chem Soc 128:3918–3919
Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R (2006b) 13C-detected protonless NMR spectroscopy of proteins in solution. Progr NMR Spectrosc 48:25–45
Bertini I, Duma L, Felli IC, Fey M, Luchinat C, Pierattelli R, Vasos PR (2004a) A heteronuclear direct detection NMR experiment for protein backbone assignment. Angew Chem Int Ed 43:2257–2259
Bertini I, Felli IC, Kümmerle R, Luchinat C, Pierattelli R (2004b) 13C–13C NOESY: a constructive use of 13C–13C spin-diffusion. J Biomol NMR 30:245–251
Bertini I, Felli IC, Kümmerle R, Moskau D, Pierattelli R (2004c) 13C–13 C NOESY: an attractive alternative to study large macromolecules. J Am Chem Soc 126:464–465
Bertini I, Fragai M, Luchinat C, Parigi G (2000) 1H NMRD profiles of diamagnetic proteins: a model-free analysis. Magn Reson Chem 38:543–550
Brüschweiler R and Ernst RR (1992) Molecular dynamics monitored by cross-correlated cross relaxation of spins quantized along orthogonal axes. J Chem Phys 96:1758–1766
Dalvit C (1992) 1H to 15N polarization transfer via 1H chemical-shift anisotropy-1H–15N dipole dipole cross corelation. J Magn Reson 97:645–650
Fernández C, Wider G (2003) TROSY in NMR studies of the structure and function of large biological macromolecules. Curr Opin Struct Biol 13:570–580
Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2004) Uniform and residue-specific 15N-labeling of proteins on a highly deuterated background. J Biomol NMR 29:289–297
Fischer MWF, Zeng L, Zuiderweg ERP (1996) Use of 13C–13C NOE for the assignment of NMR lines of larger labeled proteins at larger magnetic fields. J Am Chem Soc 118:12457–12458
Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2002) NMR analysis of a 900 KDa GroEL GROES complex. Nature 418:207–211
Frueh DP, Sun ZYJ, Vosburg DA, Walsh CT, Hoch JC, Wagner G (2006) Non-uniformly sampled double-TROSY hNcaNH experiments for NMR sequential assignments of large proteins. J Am Chem Soc 128:5757–5763
Ha Y, Shi DS, Small GW, Theil EC, Allewell NM (1999) Crystal structure of bullfrog M ferritin at 2.8 A resolution: analysis of subunit interactions and the binuclear metal center. J Biol Inorg Chem 4:243–256
Horst R, Wider G, Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2006) Proton–proton Overhauser NMR spectroscopy with polypeptide chains in large structures. Proc Natl Acad Sci USA 103:15445–15450
Kaptein R, Zuiderweg ERP, Scheek RM, Boelens R, van Gunsteren WF (1985) A protein structure from nuclear magnetic resonance data: lac Repressor headpiece. J Mol Biol 182:179–182
Liu X, Theil EC (2005) Ferritins: dynamic management of biological iron and oxygen chemistry. Acc Chem Res 38:167–175
Miclet E, Williams Jr DC, Clore GM, Bryce DL, Boisbouvier J, Bax A (2004) Relaxation-optimized NMR spectroscopy of methylene groups in proteins and nucleic acids. J Am Chem Soc 126: 10560–10570
Neuhaus D, Williamson M (1989) The nuclear Overhauser effect in structural and conformational analysis. VCH, New York
Pervushin K, Riek R, Wider G, Wüthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371
Riek R, Wider G, Pervushin K, Wüthrich K (1999) Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proc Natl Acad Sci USA 96:4918–4923
Trikha J, Theil EC, Allewell NM (1995) High resolution crystal structures of amphibian red-cell L ferritin: potential roles for structural plasticity and solvation in function. J Mol Biol 248:949–967
Tugarinov V, Choy WY, Orekhov VY, Kay LE (2005a) Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc Natl Acad Sci USA 102: 622–627
Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE (2003) Cross-correlated relaxation enhanced 1H–13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc 125:10420–10428
Tugarinov V, Ollerenshaw JE, Kay LE (2005b) Probing side-chain dynamics in high molecular weight proteins by deuterium NMR spin relaxation. An application to a 82-kDa enzyme. J Am Chem Soc 127:8214–8225
Wider G (2005) NMR techniques used with very large biological macromolecules in solution. Methods Enzymol 394:382–398
Williamson MP, Havel TF, Wüthrich K (1985) Solution Conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J Mol Biol 182:295–315
Acknowledgements
The work was supported in part by MIUR (COFIN 2005), NIH grant DK20251 (ECT and MM) and the CHRCO Foundation (ECT). The authors are grateful to Dr. Xiaofeng Liu for advice on recombinant ferritin structure/function and expression and to Dr. Rainer Kümmerle for the competent insights on spectral acquisition.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Matzapetakis, M., Turano, P., Theil, E.C. et al. 13C–13C NOESY spectra of a 480 kDa protein: solution NMR of ferritin. J Biomol NMR 38, 237–242 (2007). https://doi.org/10.1007/s10858-007-9163-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-007-9163-9