
Abstract
A pulse sequence is described for recording single-quantum 13C-methyl relaxation dispersion profiles of 13C-selectively labeled methyl groups in proteins that offers significant improvements in sensitivity relative to existing approaches where initial magnetization derives from 13C polarization. Sensitivity gains in the new experiment are achieved by making use of polarization from 1H spins and 1H → 13C → 1H type magnetization transfers. Its utility has been established by applications involving three different protein systems ranging in molecular weight from 8 to 28 kDa, produced using a number of different selective labeling approaches. In all cases exchange parameters from both 13C→1H and 1H → 13C → 1H classes of experiment are in good agreement, with gains in sensitivity of between 1.7 and 4-fold realized using the new scheme.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Solution NMR spectroscopy is a very powerful tool for the study of protein dynamics ranging over many orders of magnitude (Ishima and Torchia 2000; Palmer et al. 1996). One emerging area focuses on millisecond (ms) time-scale motions that play important roles in enzyme function (Eisenmesser et al. 2005), ligand binding (Mulder et al. 2001a; Popovych et al. 2006), molecular recognition processes (Gryk et al. 1996; Kalodimos et al. 2004) and protein folding (Hill et al. 2000; Korzhnev et al. 2004c). For these studies Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments are extremely powerful because information about both the time-scale and the thermodynamics of the process can be obtained, along with structural data in the form of chemical shift differences between the interconverting states (Korzhnev et al. 2004c). Initial protein applications focused on 15N-based CPMG dispersion measurements (Loria et al. 1999; Tollinger et al. 2001) and subsequently applications involving 1HN (Ishima and Torchia, 2003), 13Cα (Hill et al. 2000) and methyl-13C (Skrynnikov et al. 2001) spin-probes have emerged. Central to the CPMG-class of experiment has been the development by Loria and Palmer of a scheme where the influence of relaxation contributions from external protons to the spin-system probe in question can be rendered independent of the CPMG pulse rate so that the dispersion experiments report faithfully on the exchange process of interest and not on some nuance of spin-physics that complicates the analysis (Loria et al. 1999).
Of the experiments that are available for the study of ms time-scale dynamics, those that exploit methyl groups as reporters are the most sensitive (Skrynnikov et al. 2001). In this regard we have shown recently that provided that a suitable labeling scheme is employed where methyl groups are protonated in a highly deuterated protein background, methyl-TROSY multiple-quantum based CPMG relaxation dispersion data sets (Korzhnev et al. 2004a) can be obtained for systems as large as the 670 kDa proteasome (Sprangers and Kay 2007). Our original efforts in the development of methyl-13C single-quantum dispersion experiments led to schemes where magnetization originates on 13C, with sensitivity enhancement via the substantial 1H-13C steady state NOE (referred to in what follows as 13C → 1H-CPMG) (Skrynnikov et al. 2001). As our interest in studies of larger proteins increases there is a need to revisit this experiment in the hopes of substantially improving sensitivity and an obvious place to start is to consider a simple scheme with magnetization originating on 1H (1H → 13C → 1H-CPMG). Here we compare 13C → 1H-CPMG and 1H → 13C → 1H-CPMG experiments performed on a variety of different proteins, with molecular weights between 8 and 28 kDa, and with methyl labeling obtained using a number of different schemes. It is shown that very similar exchange parameters are obtained using both approaches, with gains in sensitivity for the 1H → 13C → 1H experiment ranging between 1.7 and 4-fold in the systems examined. It thus becomes possible to extend 13C single-quantum CPMG studies of ms dynamics processes to significantly larger systems, further increasing the utility of this technique.
Materials and methods
Protein samples
A uniformly 15N, selectively 13C-labeled FF domain from the human protein FBP11 was expressed in K-MOPS medium (Neidhardt et al. 1974), using Escherichia coli JM109 (DE3) cells (Promega, Madison, WI, USA), with 15NH4Cl and [1-13C]-glucose (Cambridge Isotope Laboratories, Andover, MA, USA) as the only nitrogen and carbon sources. Purification was as discussed previously (Allen et al. 2002; Jemth et al. 2005). As will be described in a subsequent publication this led to ∼ 50% 13C enrichment for methyl groups without 13C label at adjacent positions [Ala, Met, Val, Leu, Ile(γ2)], with the exception of Ile(δ1) and Thr(γ2) methyls where enrichment was on the order of 10% and scalar coupling to the adjacent carbon was noted; these residues were excluded from the analysis. A 1.0 mM protein sample was used for all experiments and the buffer conditions were: 50 mM sodium acetate, 100 mM NaCl, 0.05% NaN3, 0.2 mM EDTA, pH 5.7, 10% D2O.
The expression of both cysteine-free lysozyme (C54T/C97A; referred to in what follows as TA) and TA with an additional L99A mutation (referred to as L99A) has been described previously (Mulder et al. 2002). Expression was achieved using 13CH3-pyruvate and 15NH4Cl as the sole carbon and nitrogen sources. As has been discussed by Mulder et al. (2002) this scheme produces 13C-labeled methyl probes for Met, Leu, Val, Thr and Ile(γ2); Ala methyl groups are also enriched, however, one bond 13C(α)-13C(β) couplings were noted for a substantial fraction of these residues. NMR samples were 1.0 (L99A) or 2.0 mM (TA) protein in 50 mM Pi, 25 mM NaCl, pH 5.5, 5% D2O.
A [U-15N,12C,2H], {Ile δ1(13CH3), Leu(13CH3,13CH3), Val(13CH3,13CH3)}-labeled sample of E. coli general NAD(P)H:flavin oxidoreductase (FRE) in complex with flavin adenine dinucleotide (FAD) was prepared, as described previously (Vallurupalli and Kay 2006), using 2H-glucose supplemented with the appropriate 13C-methyl labeled precursors (Goto et al. 1999). The final sample concentration was approximately 1.5 mM in protein, 20 mM potassium phosphate, 5 mM FAD, 5 mM DTT, 0.1 mM EDTA, pH 7.4. A second fully protonated FRE-FAD complex was prepared using [1-13C]-glucose as the carbon source, with a concentration very similar to the first sample.
NMR spectroscopy
Relaxation dispersion profiles were recorded for all protein samples using both 500 and 800 MHz spectrometers, equipped with room temperature triple resonance probe-heads. 13C → 1H-CPMG data sets were recorded using the pulse scheme of Fig. 1 of Mulder et al. (2002), while 1H → 13C → 1H spectra were obtained using the sequence of Fig. 1 of the present paper. Data sets were recorded for both FF (30°C, 5°C) and lysozyme samples (25°C) with T = 30 ms (see Fig. 1) and with νCPMG values that varied between 66 Hz and 1 kHz. Each of the 13C → 1H and 1H → 13C → 1H spectra were recorded with 16 and 8 scans, respectively, with repetition delays of 1.5 s and with 64 increments in the indirect dimension, to give net acquisition times/spectrum of 0.9 and 0.4 h. Dispersion profiles of the FRE-FAD complex were obtained from spectra recorded at 800 MHz with T = 22 ms (17 and 22°C) and at 500 MHz with T = 24 and 26 ms at 17 and 22°C, respectively, along with νCPMG values between 80 − 1,000 Hz. Data sets with 16 (13C → 1H) and 8 (1H → 13C → 1H) scans were recorded along with repetition delays of 1.6 s and 64 increments in t1; net acquisition times were 0.9 and 0.4 h/spectrum.

Pulse scheme for the measurement of methyl 13C single-quantum relaxation dispersion profiles in proteins. 13C labeling need not be restricted to methyl groups, but labeling at carbon sites adjacent to methyl groups must be avoided. All narrow (wide) pulses are applied with flip angles of 90° (180°) degrees along the x-axis unless indicated otherwise. The CPMGY [CPMGX] element is of the form (δ 180y δ) n [(δ 180x δ) n ] where n is even. In cases where labeling is not restricted to the methyl positions, refocusing pulses are of the RE-BURP variety (Geen and Freeman 1991) and are applied so as to minimize excitation at 58 ppm, the center of the Leu Cα region. In this way evolution due to the three bond Cα-Cδ scalar coupling is refocused as has been described previously (Mulder et al. 2002). RE-BURP pulses are applied with durations of 500 (centered at 2 ppm) and 350 μs (7.6 ppm) at 500 and 800 MHz, respectively; alternatively a 500 μs pulse can also be applied for applications at 800 MHz centered at 22.8 ppm. In contrast, when labeling is restricted to the methyl positions rectangular pulses are employed during the CPMG pulse trains (17 kHz). All other 13C pulses are applied with the highest power possible, with WALTZ-16 decoupling (Shaka et al. 1983) achieved with a 2 kHz field. All 1H pulses are applied at high power with the exception of the water-selective pulses of the WATERGATE element (Piotto et al. 1992) at the conclusion of the pulse scheme. Between points a and e the 1H carrier is placed in the center of the methyl region; at other positions in the scheme the carrier is on water. The delays τa and τb were set to 1.8 and 1.98 ms, respectively. The phase cycle is φ1 = x, −x, φ2 = x, x, −x, −x and ψrec = x, −x, −x, x, with quadrature detection achieved by States-TPPI of φ2 (Marion et al. 1989). The durations and strengths of gradient pulses (ms, G/cm) are: g1 = (0.5 ms, 4 G/cm), g2 = (0.3 ms, 5 G/cm), g3 = (1 ms, 25 G/cm), g4 = (0.3 ms, 12 G/cm), g5 = (0.5 ms, 8 G/cm), g6 = (0.3 ms, −10 G/cm), g7 = (0.7 ms, 8 G/cm) and g8 = (0.3 ms, −20 G/cm). The inset shows a modification to the basic pulse scheme where 13C chemical shifts are recorded when the coherence of interest is of the multiple-quantum variety that offers some benefits in applications to methyl-protonated, highly deuterated proteins of high-molecular weight (not used in any of the applications here)
Samples produced using either [1-13C]-glucose or 13CH3-pyruvate contain 13C label at positions remote from methyl groups and the evolution of magnetization due to small homonuclear 13C-13C scalar couplings (in particular the three-bond coupling connecting Cδ and Cα in Leu residues) can lead to artificial dispersion profiles (Mulder et al. 2002). In applications involving proteins produced using either of these carbon sources dispersion experiments were recorded that employ selective refocusing pulses (Geen and Freeman 1991) during the T delay (Fig. 1) with carrier placement such that a null is obtained at the Cα resonance positions of Leu (Mulder et al. 2002). As described in the legend to Fig. 1 these pulses typically have the RE-BURP profile (Geen and Freeman 1991) and can be up to 500 μs in duration. This restricts the maximum νCPMG value that can be employed to approximately 1 kHz. A discussion of the use of CPMG pulse trains with RE-BURP refocusing pulses, including numerical simulations that establish that such pulses do not interfere with the extraction of accurate exchange parameters has been presented previously (Mulder et al. 2002).
Relative sensitivities of 13C → 1H and 1H → 13C → 1H methyl relaxation dispersion experiments were obtained by quantifying ratios of corresponding peaks in 2D correlation maps recorded with (i) T = 0 and with (ii) protein dependent values of T listed above and maximum νCPMG values used in each dispersion profile (∼1 kHz).
Data analysis
All data sets were processed using the nmrPipe/nmrDraw suite of programs (Delaglio et al. 1995) and analyzed as described in a series of papers (Korzhnev et al. 2004c; Mulder et al. 2002; Skrynnikov et al. 2001). Errors in peak intensities, subsequently propagated to errors in effective relaxation rates, R 2,eff(νCPMG), (see below) were estimated from duplicate experiments; a minimum error of 2% was imposed on R 2,eff(νCPMG). Dispersion profiles were fitted to a model which assumes a two-state exchange process using software written in-house that is available upon request, with errors in the model parameters (see below) estimated using the covariance matrix approach (Press et al. 1988).
Results and discussion
Figure 1 illustrates the pulse sequence used to record 13C methyl single-quantum CPMG relaxation dispersion profiles, with 1H → 13C → 1H transfer. The sequence follows directly from our previous version that begins with 13C polarization (Skrynnikov et al. 2001), with the exception of the INEPT magnetization transfer element at the start. Central to scheme is the constant time period between points a−d during which a variable number of 13C refocusing pulses is applied. These pulses can be non-selective in the case where 13C label is only restricted to the methyl carbon positions, or alternatively, selective pulses can be employed to refocus small three-bond 13C-13C homonuclear couplings when carbon sources such as 13CH3-pyruvate or [1-13C]-glucose are used to produce samples (see Materials and methods).
As we have described in detail previously, the intensities of correlations obtained in 2D 13C, 1H correlation maps can be converted directly to effective decay rates, R 2,eff, according to the relation
where νCPMG = 1/(2δ) and δ is the time between refocusing pulses, T is the duration of the constant-time CPMG relaxation delay, I(νCPMG) is the intensity of a correlation recorded in spectra obtained with the CPMG elements (a to b and c to d) and I o is the intensity of the corresponding cross-peak without the CPMG train, T = 0 (Mulder et al. 2002; Skrynnikov et al. 2001). The period extending from b to c results in the interconversion between C TR ↔ 8C TR I i Z I j Z I k Z and 2C TR I i Z ↔ 4C TR I j Z I k Z , where C TR denotes in-phase 13C methyl transverse magnetization and I i Z is proportional to the z-component of methyl proton i magnetization. This so called P-element was developed originally by Loria and Palmer in their studies of chemical exchange in 15N-1H two spin systems (Loria et al. 1999), but its utility is general for both AX2 (Mulder et al. 2001b) and AX3 (Skrynnikov et al. 2001) groups, in that it ensures that flat CPMG dispersion profiles are obtained in the absence of chemical exchange, but in the presence of external proton spins that lead to differential relaxation of in-phase and anti-phase 13C magnetization components. In the absence of the P-element such differential relaxation can produce large dispersion profiles for applications involving methyl groups that are unrelated to chemical exchange as has already been shown (Skrynnikov et al. 2001).
Previous theoretical studies established that the performance of the P-element in methyl CPMG experiments is independent of the initial magnetization conditions at the start of the constant-time relaxation period, T, and 13C → 1H-CPMG experiments were presented to show that at least in the case where the initial magnetization is proportional to C TR , robust measures of exchange could be obtained (Skrynnikov et al. 2001). Building on the earlier studies we show here that experiments where polarization originates on 1H with subsequent magnetization transfer to carbon via INEPT are also robust. As a first example we have recorded both 13C →1H-CPMG and 1H → 13C → 1H-CPMG experiments on TA lysozyme (see Materials and Methods), a protein for which we were not able to detect ms dynamic processes using methyl group probes and the 13C → 1H-based experiment several years ago (Mulder et al. 2002). Clearly if the 1H → 13C → 1H-CPMG experiment is to be of general interest then flat dispersions must be obtained for TA lysozyme using this approach as well.
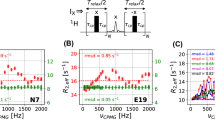
Figure 2 shows R 2,eff(νCPMG) profiles for residues V149γ1 (A), I17γ2 (B) and L99δ2 (C) recorded using 13C→1H-CPMG (bottom, o) and 1H→13C→1H-CPMG (top, x) schemes, along with best fit horizontal lines. Qualitatively, it can be seen that both approaches produce essentially flat dispersion profiles, in keeping with expectations based on our previous studies. The fits of R 2,eff(νCPMG) to flat profiles can be quantified by the relation \({RMSD=\sqrt{N^{-1}\sum\limits_i{\{R_{2,{\rm eff}}^i (\nu _{{\rm CPMG},i} )-k\}^2}}}\) where R i2,eff is the effective transverse relaxation rate at CPMG frequency v CPMG,i , R 2,eff(νCPMG) = k is the best fit horizontal line to the experimental ‘curve’ and N is the number of data points in the dispersion profile. We have selected residues with the worst (A), average (B) and best (C) RMSD values for the figure. Figure 2D plots the distribution of RMSD values for both classes of experiment, with 52 methyl groups included in the analysis. In all profiles quantified using both experiments RMSD values well under 1 s−1 were calculated. RMSDavg values of 0.5 ± 0.2 s−1 and 0.3 ± 0.1 s−1 are obtained for 13C → 1H-CPMG and 1H → 13C → 1H-CPMG experiments, respectively, with the smaller value for the 1H → 13C → 1H-CPMG data set likely due to its higher sensitivity; when experimental errors are included both values are identical.

Relaxation dispersion profiles, R 2,eff(νCPMG), for residues V149γ1 (A), I17γ2 (B) and L99δ2 (C) of TA lysozyme (25°C) recorded at a spectrometer field of 500 MHz using 13C→1H-CPMG (bottom) and 1H→13C→1H-CPMG (top) schemes, along with best fit horizontal lines. (D) Distribution of \({RMSD=\sqrt {N^{-1}\sum\limits_i {\{R_{2,{\rm eff}}^i (\nu _{{\rm CPMG},i})-k\}^2} } }\) values (13C→1H-CPMG in white; 1H→13C→1H-CPMG in grey), where R i2,eff is the effective transverse relaxation rate at CPMG frequency v CPMG,i , R 2,eff(νCPMG) = k is the best fit horizontal line to the experimental ‘curve’ and N is the number of data points in the dispersion profile
As a final note, the difference in intrinsic relaxation rates observed in both classes of experiment is striking. This can be understood, at least in a qualitative sense, by noting that the effect of the P-element is to render the evolution of magnetization during the constant-time period, T, essentially independent of the one-bond 13C-1H scalar coupling (see for example Eqs. 8–13 of Skrynnikov et al. 2001). The effective relaxation rate during T depends critically, however, on the initial conditions that vary between the two classes of experiment (C TR and 2C TR I Z for 13C → 1H and 1H → 13C → 1H transfers, respectively). It has been shown previously that contributions to the auto-relaxation rates of in-phase and anti-phase carbon magnetization from intra-methyl dipolar interactions are very different, with the auto-relaxation of 2C TR I Z considerably higher than that for C TR (Equation AI.3 of Skrynnikov et al. 2001).
Comparative studies of data sets recorded on TA lysozyme provide an important first step in establishing the validity of the pulse scheme of Fig. 1. As a second step we have compared extracted exchange parameters from fits of dispersion profiles recorded using both methods on a number of protein systems for which ms dynamics are known to be present. As described above detailed simulations and experiments provide a high level of confidence that accurate measures of exchange can be obtained from the 13C→1H-CPMG experiment and an important criteria for establishing the utility of the 1H→13C→1H approach, further to the analysis above, is to ensure the consistency of exchange values obtained from both methods.
As a first test we have used a 71-residue four helix bundle FF module from the human protein FBP11 (Allen et al. 2002) that has been prepared with 15NH4Cl and [1-13C]-glucose as the only nitrogen and carbon sources, so that exchange parameters obtained from the 13C experiments can be compared with those from the more traditional 15N-based approach as well. As described in Materials and Methods a large fraction of the methyl containing residues are labeled with 13C at methyl positions but not at the adjacent carbon position using this approach, so that a significant number of probes are available for analysis (Ala, Met, Val, Leu, Ile(γ2)). Fig. 3 shows dispersion profiles of a number of methyl groups recorded using either 13C → 1H-CPMG or 1H → 13C → 1H-CPMG experiments (30°C). Relaxation dispersion profiles recorded at 500 and 800 MHz were fit simultaneously to a global two-state model of chemical exchange \({A\mathop{\rightleftarrows}\limits_{k_B}^{k_A}B}\) and values of k ex = k A + k B and populations of the minor state, p B , are 2,290 ± 50 s−1, 2.4 ± 0.08% and 2,420 ± 50 s−1, 2.2 ± 0.06% from 13C→1H-CPMG and 1H → 13C → 1H-CPMG, respectively. Fits of 15N dispersion profiles produced values of k ex = 2,290 ± 25 s−1 and p B = 2.3 ± 0.06% that are in good agreement with results from the methyl groups.

Relaxation dispersion profiles, R 2,eff(νCPMG), for select residues from the FF domain of the human protein FBP11 recorded at 500 (lower) and 800 (upper) MHz spectrometer fields, 30°C, using either 13C → 1H-CPMG (left panel) or 1H → 13C → 1H-CPMG (right) schemes. The solid lines correspond to the best fits obtained from separate global analyses of data sets from each of the 13C → 1H-CPMG and 1H → 13C → 1H-CPMG experiments
As a final check of the consistency of the 13C-methyl data sets recorded on the FF domain sample the differences in chemical shifts between interconverting states, \({\left|\Delta \varpi \right| }\), that have been extracted by fitting dispersions from each experiment separately are plotted in Fig. 4, along with the best fit line. The small systematic deviation between data sets most likely reflects the coupling between \({\Delta \varpi}\) and p B that can occur even if exchange is not in the fast regime. Palmer and coworkers have come up with an index, α = dlnR ex/dlnΔω = 2(k ex /Δω)2/(1 + (k ex /Δω)2) (0 ≤ α ≤ 2) for quantifying the time scale of exchange (Millet et al. 2000), with α = 2 indicating fast exchange; here α values vary between 1.5 and 1.9, with an average value of 1.8 (at 500 MHz; and between 1.0 and 1.9, average of 1.5 at 800 MHz), indicating that for some but not all of the residues exchange is fast. Indeed, if p B is fixed at 2.2% for both data sets a best fit line y = 0.03 + 0.99x is obtained, while the near perfect correlation of the two data sets is retained (not shown).

Correlation of extracted chemical shift differences, \({\left|\Delta \varpi\right|}\), obtained for each residue of the FF domain with R 2,eff(67 Hz)−R 2,eff(933 Hz) > 1.8 s−1. Dispersion profiles for each type of experiment were fit globally to extract global values of (k ex , p B ) along with \({\left|\Delta \varpi \right|}\) values for each residue. The equation for the best-fit straight line is shown in the right hand corner of the plot
As a further test we have compared results from 13C → 1H-CPMG and 1H → 13C → 1H-CPMG dispersion experiments recorded on a cavity mutant of T4 lysozyme where position 99 has been mutated from Leu to Ala (referred to as L99A). L99A undergoes a single cooperative conformational transition that allows it to bind bulky hydrophobic ligands (Feher et al. 1996) despite the fact that the binding site is inaccessible from the low energy ground state according to structural data available from X-ray crystallography (Mulder et al. 2001a). The exchange process in this system has been characterized in detail previously by our group and has been found to be amenable to study by relaxation dispersion spectroscopy (Mulder et al. 2001a, 2002). Lysozyme (18.7 kDa, 164 residues) is significantly larger than the FF domain described above and experiments recorded on the L99A system allow one to establish that the good agreement between the 13C → 1H and 1H → 13C → 1H-CPMG approaches observed for the FF construct is not the result of some particular spin relaxation feature that can be attributed to the small size of the FF domain.
L99A was prepared with 13CH3-pyruvate as the carbon source (see Materials and methods) so that methyl carbons of Val, Leu, Ile(γ2) and Met are available as probes. Values of (k ex , p B ) of (1,580 ± 40 s−1, 2.8 ± 0.06%) and (1,540 ± 30 s−1, 2.7 ± 0.04%) were obtained from independent fits of 13C → 1H and 1H → 13C → 1H-CPMG data sets, respectively, that were recorded at both 500 and 800 MHz (data at the two fields were fit simultaneously in each case). Fig. 5 shows the correlation between \({\left|\Delta \varpi\right|}\) values obtained from analysis of the experiments, establishing still further that excellent agreement is obtained between the two approaches.

Correlation between \({\left|\Delta \varpi\right|}\) values obtained from analysis of dispersion profiles recorded on L99A, 25°C (see legend to Fig. 4 for details) for residues with R 2,eff(67 Hz)−R 2,eff(933 Hz) > 1.7 s−1
As a final example we consider relaxation dispersion profiles recorded on a sample of a complex between E. coli general NAD(P)H:FRE, 232 residues, 27 kDa and FAD. FRE has been shown by single molecule fluorescence to fluctuate over a range of time-scales between 10−4 and 1 s (Yang et al. 2003) and by both 13C-methyl and 15N relaxation dispersion NMR spectroscopy to undergo several distinct exchange processes with ms time-constants (Vallurupalli and Kay 2006). A highly deuterated, Ile(δ1), Leu, Val-methyl protonated sample was employed in a first set of analyses. Our previous study established that a single global analysis of the dispersion data is not appropriate (Vallurupalli and Kay 2006) and we have therefore simultaneously fit data recorded at a pair of temperatures (17 and 22°C) and at two static magnetic fields (500 and 800 MHz) on a per-residue basis. Fig. 6A compares k ex values extracted from these fits. Here values of p B and \({\Delta \varpi }\) were not constrained to be the same in each residue-specific analysis of 13C → 1H and 1H → 13C → 1H-CPMG dispersion profiles, although values of \({\Delta \varpi}\) were assumed to be temperature independent. Despite the substantial error associated with k ex values that are extracted independently for each residue it is nevertheless clear that very similar values are obtained from each of the methods. Fig. 6B plots \({\left|\Delta \varpi\right|}\) values that have been obtained from an analysis of each residue where it is assumed that k ex and p B are common to each of the 13C → 1H and 1H → 13C → 1H-CPMG profiles.

(A) Values of k ex extracted from per-residue fits of dispersion profiles recorded on a [U-15N,12C,2H], {Ile δ1(13CH3), Leu(13CH3,13CH3), Val(13CH3,13CH3)}-labeled sample of the FRE-FAD complex at 500 and 800 MHz, 17 and 22°C. Values of (k ex , p B ) were not fixed between 13C→1H-CPMG and 1H→13C→1H-CPMG experiments. (B) Correlation of \({\left|\Delta \varpi \right|}\) values obtained from separate data fits for each residue with (k ex , p B ) fixed to the same values for data sets recorded using either 13C → 1H-CPMG or 1H → 13C → 1H-CPMG schemes (see text). The equation of the best-fit straight line is shown in the lower right hand corner
In this regard it is worth noting that p B and \({\Delta \varpi}\) cannot be extracted separately from individual residue fits in many cases in FRE since for a substantial number of residues the exchange process approaches the fast regime.
The consistency between the 13C → 1H and 1H → 13C → 1H experiments is further illustrated in Fig. 7 where representative fits of dispersion profiles for Val 7 are shown. All of the profiles in the figure were fit simultaneously to common k ex and p B values but with temperature independent \({\Delta \varpi }\) values that were not fixed between the experiments. Values of \({\left|\Delta \varpi \right|}\) of 1.08 ± 0.05 and 1.01 ± 0.05 were obtained from the 13C → 1H and 1H → 13C → 1H profiles, respectively. We have also prepared a second sample of the FRE-FAD complex using [1-13C]-glucose (fully protonated) so that more methyl probes could be obtained. Studies of fully protonated samples are of interest as test cases of the methodology because the large contributions from external protons that must be ‘equalized’ for the different 13C transverse magnetization modes that evolve during T (Fig. 1) is a stringent test of the efficacy of the P-element. Dispersion profiles from both types of data sets were again shown to be consistent, as observed with the fully protonated FF domain and L99A lysozyme.

Representative fits of dispersion profiles from Val 7 of the [U-15N,12C,2H], {Ile δ1(13CH3), Leu(13CH3,13CH3), Val(13CH3,13CH3)}-labeled FRE-FAD complex; all eight dispersions were fitted simultaneously
As noted in the introduction a major motivation of the present work is to improve the sensitivity over existing experiments. It is thus critical to ascertain what gains can be achieved with schemes where polarization begins on 1H spins. A calculation of the sensitivity gain of the 1H → 13C → 1H-CPMG experiment relative to the 13C → 1H scheme that includes magnetization transfer efficiencies based on the values of the delays used in the experiments, but does not take into account relaxation during the pulse schemes, predicts a gain of a factor of 5.4. This assumes that in the 13C → 1H experiment there is a maximum NOE enhancement of the initial 13C polarization. In practice somewhat smaller gains are achieved. For example, in the case of the FF domain the average gain was 4.4 ± 0.8 (30 residues) based on spectra recorded at 30°C and T = 0 (correlation time, τC, of 4.6 ns), with an average intensity gain of 3.2 ± 0.9 at 5 °C, T = 0 (average over 30 residues; τC = 8.8 ns). Gains of 3.7 ± 1.0 and 2.4 ± 0.9 were obtained from data sets recorded at 30 (30 residues) and 5°C (30 residues), respectively, with T = 30 ms and νCPMG = 933 Hz; the smaller gains with T = 30 ms reflect the differences in intrinsic R 2,eff values that can be seen in the dispersion profiles of Fig. 3 that have been recorded with the different schemes and that have been discussed in some detail in the context of the R 2,eff profiles generated for TA lysozyme (Fig. 2). In the case of TA lysozyme gains of 3.3 ± 1.0 and 2.5 ± 1.0 are obtained in spectra recorded with T = 0 and 30 ms, respectively, at 25°C (average over 52 correlations), where τC has been measured to be 10.8 ns. Finally, a gain of 2.1 ± 0.5 is noted in experiments on the FRE-FAD complex at 17°C (τC = 18.6 ns) with T = 0 ms (average over 47 residues), that decreases to 1.7 ± 0.4 for T = 22 ms, νCPMG = 1,000 Hz (47 residues).
A number of factors can potentially contribute to lower gains in sensitivity than what might naively be expected based on simple calculations. In principle differences in methyl 13C and 1H longitudinal relaxation times would influence relative sensitivities since polarization derives from 13C and 1H spins in 13C → 1H and 1H → 13C → 1H experiments, respectively. Such differences are, however, very small. For example, average 13C and 1H T1 values of 0.31 ± 0.1 and 0.34 ± 0.1, respectively, have been calculated for methyl groups in protein L (5°C) using methyl dynamics parameters measured from 2H relaxation experiments. We have quantified such rates experimentally for the FF domain with T1(13C)/T1(1H) = 0.94 ± 0.2, 0.81 ± 0.3 at 30, 5°C; these small differences in longitudinal relaxation times have essentially no impact on deviations in relative sensitivity between the two classes of dispersion experiment nor does the temperature variation in T1 times account for the decrease in relative sensitivities with molecular size or rotational correlation time. In contrast, transverse relaxation effects would be expected to play a major role in lowering the sensitivity gain. It has previously been shown that the quality of 1H → 13C → 1H HSQC-based spectra deteriorate in applications to proteins of increasing size since the slow and fast relaxing 1H transitions that give rise to methyl spectra are interconverted by the 90° 1H pulses in the sequence (Tugarinov et al. 2003). Thus, only a small fraction of the original slowly relaxing magnetization remains in this ‘state’ for the duration of the total pulse scheme. Of course, in applications involving small proteins the difference between fast and slowly relaxing transitions is less pronounced so that the interconversion is less ‘costly’ and the sensitivity gains approach those predicted in the absence of relaxation. It is clear from the present work that for proteins with molecular weights up to at least 30 kDa substantial gains in sensitivity can be realized starting with polarization on 1H spins, although not what might have been expected at first glance.
In order to best evaluate what approach is optimal for a given application we recommend that individual 1H → 13C → 1H and 13C → 1H spectra be recorded using values of T similar to what will be used in dispersion experiments and that uncertainties in R 2,eff(νCPMG), ΔR 2,eff(νCPMG) = 1/T|ΔI(νCPMG)/I(νCPMG)|, be calculated on a per-residue basis, where ΔI is the uncertainty in a given peak intensity (see Eq. 1; the error in I o is assumed to be negligible since the intensities of correlations are much larger without the constant-time element). In the absence of systematic errors and assuming that very similar T values are selected, as would normally be the case, the experiment producing the highest sensitivity spectra should be chosen, not surprisingly, since these spectra will have the lowest uncertainties in R 2,eff(νCPMG). Thus, so long as I(νcpmg) values are larger in 1H → 13C → 1H spectra uncertainties in rates will be smaller and 1H polarization experiments will be more sensitive monitors of exchange processes. Of course, this increased sensitivity can be ‘traded’ for larger T values that are advantageous in studying slower exchange events. For example, suppose that intrinsic effective relaxation rates are 30 and 20 s−1 for 1H → 13C → 1H and 13C → 1H dispersions, respectively (see Fig. 2), corresponding to the largest (and most different) rates that have been quantified in the examples considered in this work and that the ratio of sensitivities of 1H → 13C → 1H and 13C → 1H spectra (T = 0; without the constant-time element) is 3:1 (as is the case for TA lysozyme). It is straightforward to show that similar intensities in both classes of spectra will be obtained for T values of 57 (1H → 13C → 1H) and 30 (13C → 1H) ms, so that minimum νCPMG values of 35 and 67 Hz can be employed in each case. Finally, although the difference in intrinsic rates of magnetization considered in this example is substantial it still remains the case that the 1H → 13C → 1H experiment is the more sensitive of the two for values of T up to 110 ms, values that are much larger than one would likely ever use in studies of proteins. Thus, at least for typical relaxation rates measured here, it is hard to imagine cases involving small to medium sized proteins, similar to those surveyed presently, where the 1H → 13C → 1H scheme would not be the one of choice.
Of course, for applications to very high molecular weight proteins, where the losses are most severe, it is likely that both experiments will fail. A methyl-TROSY multiple-quantum CPMG scheme has been developed for these cases (Korzhnev et al. 2004b), where magnetization originating from the slowly relaxing 1H transitions is preserved throughout the course of the sequence, resulting in large gains in sensitivity. Because multiple-quantum and not single-quantum coherences are relevant in this experiment dispersion profiles are sensitive to changes in both 1H and 13C chemical shifts between exchanging sites that offers both advantages and disadvantages relative to the single-quantum variant (Korzhnev et al. 2004b).
In summary we have presented an improved pulse scheme for the measurement of 13C-methyl single-quantum relaxation dispersion profiles. The experiment has been tested on a number of different proteins with molecular weights ranging from 8 to 28 kDa that have been methyl-labeled using a variety of different protocols. In all cases excellent agreement between exchange parameters calculated from dispersions measured using 13C→1H and 1H→13C → 1H based experiments is obtained, with gains in sensitivity ranging from 1.7 to 4-fold. It is clear that the new 1H→13C→1H-CPMG sequence will extend the utility of single-quantum methyl dispersion spectroscopy to studies of larger protein systems than would be otherwise possible, or alternatively, in cases where both 13C→1H and 1H→13C→1H approaches are feasible that the new method will facilitate extraction of exchange parameters with increased accuracy.
References
Allen M, Friedler A, Schon O, Bycroft M (2002) The structure of an FF domain from human HYPA/FBP11. J Mol Biol 323:411–416
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRpipe—a multidimensional spectral processing system based on unix pipes. J Biomol NMR 6:277–293
Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D (2005) Intrinsic dynamics of an enzyme underlines catalysis. Nature 438:117–121
Feher VA, Baldwin EP, Dahlquist FW (1996) Access of ligands to cavities within the core of a protein is rapid. Nat Struct Biol 3:516–521
Geen H, Freeman R (1991) Band-selective radiofrequency pulses. J Magn Reson 93:93–141
Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE (1999) A robust and costeffective method for the production of Val, Leu, Ile (δ1) methyl-protonated N-15-, C-13-, H-2-labeled proteins. J Biomol NMR 13:369–374
Gryk MR, Jardetzky O, Klig LS, Yanofsky C (1996) Flexibility of DNA binding domain of trp repressor required for recognition of different operator sequences. Protein Sci 5:1195–1197
Hill RB, Bracken C, DeGrado WF, Palmer AG (2000) Molecular motions and protein folding: characterization of the backbone dynamics and folding equilibrium of αD-2 using C-13 NMR spin relaxation. J Am Chem Soc 122:11610–11619
Ishima R, Torchia DA (2000) Protein dynamics from NMR. Nat Struct Biol 7:740–743
Ishima R, Torchia DA (2003) Extending the range of amide proton relaxation dispersion experiments in proteins using a constant-time relaxation-compensated CPMG approach. J Biomol NMR 25:243–248
Jemth P, Day R, Gianni S, Khan F, Allen M, Daggett V, Fersht AR (2005) The structure of the major transition state for folding of an FF domain from experiment and simulation. J Mol Biol 350:363–378
Kalodimos CG, Biris N, Bonvin AMJJ, Levandoski MM, Guennuegues M, Boelens R, Kaptein R (2004) Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305:386–389
Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE (2004a) Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J Am Chem Soc 126:3964–3973
Korzhnev DM, Kloiber K, Kay LE (2004b) Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J Am Chem Soc 126:7320–7329
Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE (2004c) Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature 430:586–590
Loria JP, Rance M, Palmer AG (1999) A relaxation-compensated Carr–Purcell Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc 121:2331–2332
Marion D, Ikura M, Tschudin R, Bax A (1989) Rapid recording of 2D NMR-spectra without phase cycling—application to the study of hydrogen-exchange in proteins. J Magn Reson 85:393–399
Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG (2000) The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc 122:2867–2877
Mulder FAA, Hon B, Mittermaier A, Dahlquist FW, Kay LE (2002) Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J Am Chem Soc 124:1443–1451
Mulder FAA, Mittermaier A, Hon B, Dahlquist FW, Kay LE (2001a) Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol 8:932–935
Mulder FAA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE (2001b) Measurement of slow (μs-ms) time scale dynamics in protein side chains by N-15 relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc 123:967–975
Neidhardt FC, Bloch PL, Smith DF (1974) Culture Medium for Enterobacteria. J Bacteriol 119:736–747
Palmer AG, Williams J, McDermott A (1996) Nuclear magnetic resonance studies of biopolymer dynamics. J Phys Chem 100:13293–13310
Piotto M, Saudek V, Sklenar V (1992) Gradient-tailored excitation for single-quantum NMR-spectroscopy of aqueous solutions. J Biomol NMR 2:661–665
Popovych N, Sun SJ, Ebright RH, Kalodimos CG (2006) Dynamically driven protein allostery. Nat Struct Mol Biol 13:831–838
Press WH, Flannery BP, Teukolsky SA, Vetterling WT (1988) Numerical Recipes in C. Cambridge University Press, Cambridge
Shaka AJ, Keeler J, Frenkiel T, Freeman R (1983) An improved sequence for broad band decoupling-WALTZ-16. J Magn Reson 52:335–338
Skrynnikov NR, Mulder FAA, Hon B, Dahlquist FW, Kay LE (2001) Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J Am Chem Soc 123:4556–4566
Sprangers R, Kay LE (2007) Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 445:718–722
Tollinger M, Skrynnikov NR, Mulder FAA, Forman-Kay JD, Kay LE (2001) Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc 123:11341–11352
Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE (2003) Cross-correlated relaxation enhanced H-1-C-13 NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc 125:10420–10428
Vallurupalli P, Kay LE (2006) Complementarity of ensemble and single-molecule measures of protein motion: a relaxation dispersion NMR study of an enzyme complex. Proc Natl Acad Sci USA 103:11910–11915
Yang H, Luo GB, Karnchanaphanurach P, Louie TM, Rech I, Cova S, Xun LY, Xie XS (2003) Protein conformational dynamics probed by single-molecule electron transfer. Science 302:262–266
Acknowledgments
P. L. and P. V. are supported by fellowships from the Hellmuth Hertz foundation and the Canadian Institutes of Health Research (CIHR) Training Grant in Protein Folding and Disease. This research was supported by a grant from the CIHR. L.E.K. holds a Canada Research Chair in Biochemistry.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lundström, P., Vallurupalli, P., Religa, T.L. et al. A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity. J Biomol NMR 38, 79–88 (2007). https://doi.org/10.1007/s10858-007-9149-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-007-9149-7