
Abstract
A new tumor-targeted drug-loading material, the amphiphilic peptide DGRGGGAAAA (P10) was designed and synthesized, and its self-assembly behavior, drug-loading effects and in vitro characteristics were studied. P10 was synthesized by solid-state synthesis and doxorubicin (DOX) was loaded via dialysis. P10 and DOX were mixed with a mass ratio of 6:1 to form regular round spheres. The interconnection between groups was analyzed spectroscopically and the sphere morphology was studied with SEM and a zeta particle size analyzer. Fluorescence spectroscopy was used to analyze the ability of P10 to form micelles and the efficiency of micelle entrapment, and the drug-loading ratio and drug release characteristics were detected. Finally, the in vitro antitumor activity of P10 was studied with HeLa cells as a model. The results showed that P10’s critical micelle concentration (CMC) value and its average grain diameter were approximately 0.045 mg/L and 500 nm. The micelle entrapment ratio and drug-loading ratio were 23.011 ± 2.88 and 10.125 ± 2.62%, respectively, and the in vitro drug-releasing properties of P10 were described by the Zero-order model and the Ritger-Peppas model. Compared with DOX, P10-DOX had a higher tumor cell inhibition ratio and a dose-effect relationship with concentration. When P10-DOX’s concentration was 20 μg/mL, the inhibition ratio was 44.17%. The new amphiphilic peptide designed and prepared in this study could be a tumor-targeted drug-loading material with better prospects for application.

In this paper, a new tumor-targeted drug-loading material, the amphiphilic peptide DGRGGGAAAA (P10) is designed and synthesized, and its self-assembly behavior, drug-loading effects and in vitro characteristics are studied, providing a theoretical basis and design ideas for further studies and the development of targeted drug-loading materials on tumor cells.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cancers are common diseases that are seriously life-threatening, and chemotherapy is one of the main clinical treatments for cancers. Chemotherapeutic drugs destroy a great number of normal cells as well as kill cancer cells. Toxic side effects, poor compliance and control of metastasis are the negative outcomes of chemotherapy [1, 2]. Studies have found that certain amphiphilic polymers can catch and transport hydrophobic drugs with the help of their hydrophobic cores preventing the absorption of proteins and protecting the drugs from being eliminated by blood [3, 4]. For example, membrane-permeable poly(ethylene glycol)-poly(dl-lactide)(PEG-PLA) block copolymer micelles can transport drugs by self-assembly, forming drug delivery carriers, and this technique has been widely applied to drug-delivery [5, 6]. However, since the carrier materials lack targetability, they can not accumulate on tumor sites effectively, and with a large molecular weight, they present problems of poor degradation in vivo and low clinical efficacy.
In recent years, studies have found that with the special physiological and pathological structure of tumor tissues, nanoparticle preparations can strengthen the active target effect of the drug carriers, taking advantage of the recognition reaction, specificity and overexpression of the receptors of tumor cells, thus apparently improving pharmaceutical preparations’ targetability and curative effects as well as decreasing the detoxification and side effects. RGD is a small peptide containing the Arg-Gly-Asp sequence, which exists widely in organisms, and is the reaction recognition site of integrin and its ligand. αvβ3, a type of glycoprotein adhesion molecule expressed on the cell surface, is an important member of the integrin family. In tumor cells, such as melanoma, glioblastoma and ovarian cancer, αvβ3 is overexpressed [7,8,9]. Exogenous RGD, as a specific ligand of integrin αvβ3, can target the effective link-coupled antitumor molecules into tumor sites, reducing the destruction of normal tissue cells in the process of treatment and effectively improving the drug’s inhibitory rate on tumor cells [5, 10, 11].
Polypeptides are a new biomedical material with wide prospects for application [12,13,14]. Zhang et al. designed and synthesized a series of small molecular surfactant amphiphilic polypeptides X6Kn (X is leucine, alanine or valine; n = 1–5) [15]. A6K,V6K2 and L6K3 self-assembled to form nanotube structures at the concentration of 1.0 mg mL−1. Xu et al. changed the polypeptide molecular geometrical configuration by changing the number (m = 3, 6, 9) of Ala residues in the AmK polypeptide [16]. Studies showed that the structures of this type of surfactant active peptides were similar to those of natural phospholipid molecules with excellent integration. However, since hydrophobic portions exist in hydrophobic amino acids, it strengthens the affinity of the erythrocyte membrane and heamolyticus is improved when the peptide chain loads hydrophobic drugs [17, 18]. Therefore, without changing the charges and activity, reducing the hydrophobic interaction is one factor to consider. When the carbon number in the alkyl chain is less than 10, with the decrease in the hydrophobic interaction between the alkyl chains, the polypeptide molecular chain can not assemble [16, 19]; thus, a peptide chain with bonds is needed. The middle peptide should have features such as good flexibility, maximum degrees of freedom of the peptide frame and sufficient space at the hydrophobic sides to catch drugs. The circular sequence (GGGGS)n (n ≤ 6) proposed by Huston et al. was designed as an excellent connecting peptide and had been widely applied to the construction of fusion proteins [20,21,22,23].
Based on the abovementioned background, we designed an amphiphilic peptide chain DGRGGGAAAA (P10) and studied the loading effect on Adriamycin, the in vitro release curve and the critical micelle concentration (CMC). Relative mathematical models were used to assess the micelle’s release mechanics, and HeLa cells were used to carry out cytotoxicity and cell uptake experiments to assess the targeted therapy effects on tumor cells, providing a theoretical basis and design ideas for further studies and the development of targeted drug-loading materials on tumor cells.
2 Materials and methods
2.1 Materials and main reagents
Doxorubicin hydrochloride analytical reagent: Najing Dilger Medical Technology Co. Ltd.; amino acid, 2-chlorotrityl chloride resin, 1-hydroxybenzotriazole (HOBT): GL Biochem (Shanghai) Ltd.; methanol, piperidine, dichloromethane (DCM), dimethylformamide (DMF), ethylether, trifluoroacetic (TFA), triethylamine analytical reagent: Sinopharm Chemical Reagent Co. Ltd; N,N-diisopropylcarbodiimide (DIC): Suzhou Highfine Biotech Co. Ltd.; pancreatin: Sigma; culture media: Gibco Company; HeLa cells: Cell Bank of the Chinese Academy of Science.
2.2 The synthesis of the linear peptide P10
P10 was synthesized by solid-state synthesis with the amino acids protected by Fmoc as monomers and DIC as catalysts [24, 25]. First, 1.0 g 1.03 mmol/g 2-chlorotrityl chloride resin was added to 15 mL DCM to swell for 3 min, and 1.5 mL DIEA was added to a 15 mL DMF solution containing 0.55 g Fmoc-Asp (OtBu)-OH, 0.45 g HOBT and 1.5 mL DIC to prepare a coupling solution. The coupling solution was added into 2-chlorotrityl chloride resin to react for 1.5 h. The Kaiser experiment was performed to prove that they completely coupled. The unreacted active chlorine group was sealed with methanol and DCM, and they reacted for 30 min. After the solution was washed with DMF several times, Fmoc at the amino end of the peptide was removed with 20% piperidine, and they reacted for 20 min. The Kaiser experiment was carried out to prove the loss of -NH2. The resin was washed several times with 15 mL DMF. After fully washed, the next amino acid was added until P10’s last amino acid, Fmoc-Ala-OH. Protective groups in the coupling compound were washed and removed with 20% piperidine for 30 min, and the coupling compound was washed with 20 mL CH3OH for 5 min. TFA: water (volume ratio is 95:5). The resin was shaken for 2 h. The polypeptide was removed from the resin, the side-chain of the polypeptide as deprotected, and the crude product was achieved after being deposited in ice-cold ethylether.
2.3 Purification of P10 with preparative HPLC
The crude product of P10 described in 2.2.1 was dissolved in water. After being filtered with a 0.45 μm filter membrane, P10 was purified with RP-HPLC. Chromatographic conditions: Sephadex G-100, 5 μm, 1 × 18 cm column, Eluant A: 0.1% TFA/H2O, Eluant B: 0.1% TFA/80% acetonitrile-H2O (V/V), gradient elution 20 → 80% B, 20 min, velocity 1.0 mL/min, detection wavelength λ = 214 nm.
2.4 Mass spectrometric analysis of P10
Liquid chromatography-mass spectrometry (ESI-MS, LCMS-8030) was used to analyze P10’s molecular weight. The spectrometry conditions were as follows: ion source: ESI(+), scan target product with SIM pattern, scanning range (m/z): 800–860, temperature at the interface: 300 °C, temperature at the DL: 250 °C, temperature of the heat block: 400 °C, drying gas flow: 15 L/min, voltage at capillary: 4500 V, detection voltage: 1960 V.
2.5 Preparation of the P10-DOX nanosphere
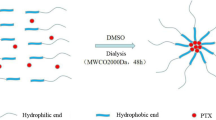
Loaded micelles were prepared via dialysis [26]. The process is shown schematically in Fig. 1. Under ultrasonic conditions, 30 mg P10, 5 mg DOX, and 0.5 mL triethylamine were added to 5 mL DMF solution and dialyzed with deionized water in a MWCO 1000-Da dialysis bag for 48 h to remove organic solvent and free DOX, forming micelles. Micelles were frozen and dried for use.

Sketch of the formation of the P10-DOX nanosphere
2.6 Detection of the characterization and particle size of P10-DOX nanospheres
P10-DOX powder was placed on a slide glass with dual adhesive tape to make a SEM samples. After metal spraying, SEM (S-4800, Hitachi Limited) was used to observe its morphology. A certain amount of P10-DOX powder was dispersed in deionized water, and a Mastersizer LPSA (Mastersizer 3000, UK) was used to detect its particle size and distribution.
2.7 Detection of the entrapment efficiency and drug-loading ratio of P10-DOX
The entrapment efficiency and drug-loading ratio of P10-DOX were detected according to formula (1) and (2) [27]:
where We is the amount of drugs in micelle; Wt is the initial amount of drugs; Wm is the total mass of micelles.
2.8 Analysis on P10-DOX nanospheres with Fourier transformation infrared spectroscopy and fluorescence spectrometry
A Fourier transformation infrared spectrometer (IRPrestige-21, Shimadzu, Japan) was used to analyze the sample’s potassium bromide tablet. The scanning range was 300–4000 cm−1 and the resolution was 4 cm−1.
A pyrene solution (6 × 10−7 mmol/L) was prepared with acetone as a solvent. Then, 50 μL pyrene solution was added to ten 25 mL conical flasks. After acetone had completely evaporated, a 10 mL P10 dispersion ranging from 0.625 × 10−4 g/L to 0.2 g/L was added to conical flasks shielded from light for 12 h to balance pyrene. With P10’s longest ultraviolet absorption wavelength of 255 nm as the excitation wavelength, the spectrum was recorded from 290 nm to 350 nm by a fluorescence spectrophotometer (RF-5301PC, Shimadzu, Japan). The slit-width of excitation and emission was 5 nm. Fluorescence intensity (I307/I300) ratios at 307 and 300 nm were calculated, and a figure was made containing the fluorescence intensity ratios and the logarithm of copolymer concentration. The intersection was the CMC value with the ratio of I307/I300 increased obviously.
2.9 Detection release profile in vitro
In vitro release mechanics experiments were carried out with the dialysis bag diffusion method [5]. 1 mg/mL P10-DOX solution was prepared with pH = 1.2, 4.5 and 6.8 Phosphate buffer saline (PBS) buffers and placed into a dialysis bag (MWCO = 1000 Da), which placed in a flask. Then, 30 mL pH = 1.2, 4.5 and 6.8 PBS buffers were added into the flask, which was placed in a shaker away from light at 37.0 ± 0.5 °C and 100 r/min, and 2 mL PBS outside the dialysis bag was removed at different periods. An ultraviolet spectrophotometer (754PC, Shanghai APL) was used to detect its absorbance at 480 nm. Then, 2 mL dissolution medium was immediately added to the PBS. The removed solution was filtered with a microporous filter. The intensity was calculated according to its standard curve equation (Formula 3). Cumulative drug release ratio (Mt, %) at different periods was assessed according to Formula 4.
where Y is DOX hydrochloride’s absorbance (Abs) detected by an ultraviolet spectrophotometer; X is DOX hydrochloride’s intensity (μg/mL); Ve is the dissolution medium’s displaced volume (mL); V0 is the initial releasing solution volume (mL); Ci is drug intensity (μg/mL) in dissolution at the i-th replacement; n is the number of replacing dissolution; Cn is the drug intensity (μg/mL) in dissolution at the n time’s replacement; mdrug is the P10-DOX nanosphere’s quality (mg); D is the drug-loading ratio.
To confirm the dynamic type and possible mechanism of DOX hydrochloride’s release from micelles, according to the P10-DOX nanosphere’s release behavior in the manually stimulated gastric juice, tumor environment and manually stimulated intestinal juice, 6 kinds of mathematical models (Zero-order, First-order, Higuchi, Hixson-Crowell, Weibull and Ritger-Peppas formula) were used to study the in vitro release mechanism.
2.10 Cytotoxicity experiment of P10-DOX nanospheres
HeLa cells were cultured according to the standard empirical method. The HeLa cell cryogenic vial was removed from a liquid nitrogen or dry ice container and was placed in a 37 °C sink for quick thawing. Then, it was placed in 2 mL complete medium containing RPMI-1640, 10% fetal bovine serum and 1% penicillin-streptomycin, and the supernatant was removed by centrifugation (800 r/min). The sedimentary cells were resuspended with the complete medium and placed into a tissue culture dish, which was put into a CO2 culture box and cultured at 37 °C and 5% CO2. The recovered HeLa cells were subcultured over three times. The following experiment was carried out when the cells were in excellent conditions. HeLa cells were incubated until 90% of the cells were in an adhered state and then digested with 1 mL 0.25% trypsin in the culture box for 2 min. Then, they were centrifuged at 800 r/min for 5 min. The sedimentary cells were resuspended with complete medium, and the number of HeLa cells was confirmed with a hemocytometer. After HeLa cells were resuspended with the complete medium, the intensity was 8 × 104/mL. Then, 100 μL cell suspension as drawn with a pipettor tube and added to a 96-well plate (approximately 8000 cells/well). It was placed into a CO2 culture box and cultured until the cells were in an adhered state. Nine concentrations (40, 20, 10, 5, 2.5, 1.25, 0.62, 0.31 and 0 μg/mL) of P10-DOX and DOX solution were added with eighteen coupling wells being designed. The cells cultured in a CO2 culture box for 24 h were observed under a microscope upside-down. After the original culture medium was drawn, approximately 100 μL cell culture medium containing 0.09% CCK-8 was added and placed into a CO2 culture box and cultured for 2 h again. Each hole’s absorbance was detected with a full-featured microplate reader (Synergy NEO, Bio Tek, USA) at 450 nm.
2.11 Cellular uptake experiment of P10-DOX nanospheres
The process in which HeLa cells were placed into a 6-well plate (2 × 105 cells/well) or a 24-well plate (5 × 104 cells/well) was the same as cytotoxicity experiment of P10-DOX nanospheres. After they were cultured until the cells were in an adhered state, P10-DOX nanospheres were added. After culture in a constant-temperature CO2 culture box for 2 and 6 h, HeLa cells were dyed with DAPI, a dye for nuclei. Laser scanning confocal microscopy (FV1200, OLYMPUS, Japan) was used to observe images.
2.12 All statistical analyses with SPSS13.0
Data were expressed as the mean and standard deviation, and differences were compared with t-tests and analysis of variance. A difference of p < 0.05 was statistically significant.
3 Results and discussion
3.1 HPLC purification and mass spectrometric analysis of polypeptide P10
HPLC was used for purification analysis on the purified product. P10’s retention time was 7.342 min and its purification was approximately 96.08%. In mass spectrometry, [M + H]+ = 802.35, [M + 2H]2+ = 803.35, which corresponded to the theoretical results. Therefore, the synthesized product was the target product P10.
3.2 Detection of the P10-DOX nanosphere’s characterization, drug-loading ratio and entrapment ratio
We analyzed the results of Fourier transform infrared spectroscopy [28,29,30,31]. In Fig. 2(a), 3280 cm−1 is νNH, 1627 cm−1 is νC=O, 1541 cm−1 is βNH, and 1340 cm−1 is νCN, thus it is a typical amide structure; 2976 cm−1 is νasCH3 alkane, with νasCH2 and νsCH2 alkane nearby, and 3080 cm−1 is νCH. There is carboxy νC–O near 1203 cm−1, and it is regarded as COOH. There is primaquine δNH2 near 1627 cm−1, and it is regarded as a hydrophobic terminal.

P10-DOX nanospheres’ infrared spectrogram
In Fig. 2(b), 3462 cm−1 is the stretching vibration peak of νO–H, 2987 cm−1 is the νC–H stretching vibration peak of DOX, 1618 cm−1 is the stretching vibration peak of νC=O, 1419 cm−1 is β=CH, 1284 cm−1 is νCN, 1209 cm−1 is νasC-O-C and 987 cm−1 is νHN. Therefore, it was confirmed that DOX existed in the compound. The results agreed with reference [31].
In Fig. 2(c), It indicated the presence of DOX that 3462 cm−1 was νO–H and the ring tension at 1652 cm−1 was νC=C. 1627, 1541, and 1203 cm−1 were νC=O, βNH, and νC–O of the hydrophilic chain of P10 peptide, respectively, indicated that P10 successfully entrapped DOX. They still existed in P10-DOX and did not offset, indicating that there was no covalent bond producing in the course of forming P10-DOX. Two peaks at 2976 and 3080 cm−1 disappeared with P10 and DOX bound, indicating the hydrophobic chain was coated in the hydrophobic core.
Scanning electron microscopy (SEM) was used to observe the morphology of the P10-DOX nanospheres. It was found that their appearance was spherical, and they were homogeneous (Fig. 3), and the average grain diameter was approximately 458 nm (Fig. 4). After dialysis, there was 7.1 mL of solution left. Then, 1 mL P10-DOX nanosphere solution was freeze-dried, and 1.6 mg powder was produced. The absorbance was 0.406. According to formula (3), P10-DOX nanospheres’ drug-loading ratio and entrapment ratio were 10.12 ± 2.62 and 23.01 ± 2.88%, respectively.

SEM image of P10-DOX nanospheres

Grain diameter analysis image of P10-DOX nanospheres
3.3 Detection of P10-DOX nanospheres’ critical micelle concentration
The P10 copolymer was made up of a hydrophobic A4 block and hydrophilic RGD block, which could form micelles by means of self-assembly in water solution. With pyrene as the hydrophobic probe, the P10-DOX nanosphere’s CMC value was detected with fluorescence spectrometry.
As shown in Fig. 5a, when the concentration was low, there was no micelle in the system and pyrene’s fluorescence intensity values stayed the same, indicating that the copolymer molecules existed in water in a dispersed state. With the increase of P10’s intensity, pyrene’s fluorescence intensity values increased, and there was a redshift in the band (300 → 307 nm), indicating the micelles began to form in the solution.

P10-DOX nanosphere’s fluorescence spectrum analysis image
As shown in Fig. 5b, I307/I300’s ratio changed slightly in the low concentration range. When the copolymer’s concentration increased over a certain point (0.25 mg/L), the ratio of I307/I300 increased obviously. These changes indicated that pyrene entered P10’s hydrophobic core to form micelles. The CMC value was calculated with the intersection of extrapolated lines, approximately 0.45 mg/L, indicating that P10 had a better ability to form nano micelles in water solution [32, 33].
3.4 Analysis on the P10-DOX nanosphere’s drug-releasing properties in vitro
In the treatment process, protein and polypeptide, as drug-loading materials, were sensitive to enzymes and difficult to penetrate the gastrointestinal mucosa. They could only be used in injections, which is terribly inconvenient [34, 35]. The World Health Organization (WHO) had issued clear definitions, standards and methods in judging drugs’ release and dissolution [36, 37]. Our study researched P10-DOX nanosphere’s cumulative amount of release in manually stimulated gastric juice (pH = 1.2), manually stimulated intestinal liquid (pH = 6.8) and manually stimulated tumor environment (pH = 4.5).
P10-DOX nanosphere’s drug-releasing mechanism in different media were shown in Fig. 6. The loading material’s drug release was dependent on pH. In the pH 6.8 medium, the 36-h and 96-h cumulative release amount was 2.31 ± 0.12 and 7.51 ± 0.38%. In the pH 4.5 medium, the 36-h and 96-h cumulative release amount was 4.03 ± 0.20 and 24.51 ± 1.23%. In the pH 1.2 medium, the 36-h and 96-h cumulative release amount was 37.79 ± 1.89 and 72.81 ± 3.64%.

Curve of P10/DOX’s drug release in vitro
Therefore, P10-DOX nanospheres were more suitable for release in gastric juice and could control drug release to some degree. Judging from the analysis of the drug release in vitro in different media, P10-DOX nanospheres’ drug-releasing behavior was responsive to pH to some extent, making it a potential sustained release formulation in gastric juice. A short time after patients took drugs, they reached the effective concentration, which maintained for long term, showing the sustained release effects.
As shown in Table 1, P10-DOX nanospheres’ drug-releasing behavior in manually stimulated gastric juice agreed with that in manually stimulated intestinal liquid, that is, the Zero-order model’s fitting correlation (R2 = 0.9432 and R2 = 0.9295) was superior to that of other fitting models. As exhibited the fitting results in Table 1, the Ritger-Peppas model’s fitting correlation (R2 = 0.9667) for P10-DOX nanospheres in a manually stimulated tumor environment was superior to that of other fitting models. It was shown that the Zero-order model and Ritger-Peppas model were closer to the experimental results, that they could better describe the relationship between the cumulative release and the time for P10-DOX nanospheres in manually stimulated gastric juice, manually stimulated intestinal liquid and a manually stimulated tumor environment. The P10-DOX nanosphere’s drug-releasing mechanism was the result of erosion and osmotic pressure on the P10 peptide chain.
3.5 Experiment on cytotoxicity and cellular uptake
As shown in Fig. 7, both naked DOX and P10-DOX nanospheres had lethal effects on tumor cells and were concentration-dependent. The cellular survivals were 52.11 and 81.02% at 5 μg/mL P10-DOX and DOX, respectively. When treated with 20 μg/mL of P10-DOX and DOX, those would decreased to 14.35 and 55.83%. With the increasment of concentration, the tumor cellular inhibition ratios increased apparently at the range of 5–20 μg/mL. The tumor cellular inhibition ratios of P10-DOX were 0.55 and 2.89 times greater than that of DOX at the concentration of 5 and 20 μg/mL, respectively, indicating that P10-DOX had better biocompatibility.

Experiments on DOX’s and P10-DOX’s cytotoxicity
To further studied the correlation between P10-DOX’s value-added activity and tumor cells’ uptake, confocal laser scanning microscopy was used to observe the internalization of DOX and P10-DOX. As shown in Fig. 8, P10-DOX’s red fluorescence (Fig. 8g, h) was stronger than DOX’s red fluorescence (Fig. 8e, f), proving that P10-DOX had a higher internalization effect on tumor cells.

Laser confocal scanning microscopies of DOX and P10-DOX
4 Conclusion
Amphiphilic polypeptide P10 was used to enclose DOX, which improved the treatment effects on tumor cells. The method was simple and efficient. P10-DOX nanospheres could achieve DOX’s directional release and sustained release and was responsive to pH to some extent. The Zero-order and the Ritger-Peppas model could better fit the relationship between the cumulative release and the time for P10-DOX nanospheres in manually stimulated gastric juice, manually stimulated intestinal liquid and a manually stimulated tumor environment. P10-DOX nanospheres’ drug-releasing mechanism was the result of erosion and osmotic pressure on the P10 peptide chain. As a drug-loading material, P10 had a medium CMC value and caused cellular uptake by combining with tumor cells’ specificity. The results showed that P10-DOX had practical significance, could facilitate an improvement in curative effects on tumor cells, and had good prospects for application. Further study on the potential for loading DOX onto target tumor cells in animals was needed.
References
Song X, Zhu JL, Wen Y, Zhao F, Zhang ZX, Li J. Thermoresponsive supramolecular micellar drug delivery system based on star-linear pseudo-block polymer consisting of β-cyclodextrin-poly (N-isopropylacrylamide) and adamantyl-poly(ethylene glycol). J Colloid Interf Sci. 2017;490:372–9.
Pilar DLP, Azab AK. Nanoparticle delivery systems, general approaches, and their implementation in multiple myeloma. Eur J Haematol. 2017;98:529–41.
Xu H, Lu X, Li J, Ding D, Wang H, Li X, Xie W. Superior antitumor effect of extremely high drug loading self-assembled paclitaxel nanofibers. Int J Pharm. 2017;526:217–24.
Raj V, Prabha G. Synthesis, characterization and in vitro drug release of cisplatin loaded Cassava starch acetate–PEG/gelatin nanocomposites. J Assoc Arab Univ Basic Appl Sci. 2016;21:10–16.
Li C, Wang W, Xi Y, Wang J, Chen JF, Yun J, Le Y. Design, preparation and characterization of cyclic RGDfK peptide modified poly(ethylene glycol)-block-poly(lactic acid) micelle for targeted delivery. Mat Sci Eng. 2016;64:303–9.
Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed. 2010;45:1198–215.
Meidell KZ, Robinson R, Vieira-de-Abreu A, Gormley AJ, Ghandehari H, Grainger DW, Campbell RA. RGDfK-functionalized gold nanorods bind only to activated platelets. J Biomed Mater Res A. 2017;105:209–17.
Zhang L, Zhu S, Qian L, Pei Y, Qiu Y, Jiang Y. RGD-modified PEG-PAMAM-DOX conjugates: in vitro and in vivo studies for glioma. Eur J Pharm Biopharm. 2011;79:232–40.
Zhao H, Wang M, Zhou P, Wang Q, Zhou ZG, Wang DL, Yang H, Yang SP. RGD-conjugated titanium dioxide nanoparticles: targeted near-infrared photothermal therapy for alpha(v)beta(3) integrin overexpressed cancer cells. J Mater Sci. 2017;52:13356–64.
Ruoslahti E. The RGD story: a personal account. Matrix Biol. 2004;22:459–65.
Wu Z, Cheng X, Hong H, Zhao X, Zhou Z. New potent and selective αvβ3 integrin ligands: Macrocyclic peptides containing RGD motif synthesized by sortase A-mediated ligation. Bioorg Medi Chem Lett. 2017;27:1911–3.
Ulijn RV, Smith AM. Designing peptide based nanomaterials. Chem Soc Rev. 2008;37:664–75.
Shivhare K, Garg C, Priyam A, Guptab AS, Kumar P. Enzyme sensitive smart inulin-dehydropeptide conjugate self-assembles into nanostructures useful for targeted delivery of ornidazole. Int J Biol Macromol. 2018;106:775–83.
Kuang H, Ku SH, Kokkoli E. The design of peptide-amphiphiles as functional ligands for liposomal anticancer drug and gene delivery. Adv Drug Deliv Rev110. 2016;111:80–101.
Santoso SS, Vauthey S, Zhang S. Structures, function and applications of amphiphilic peptides. Curr Opin Colloid In. 2003;7:262–6.
Zhang JH, Zhao YR, Han SY, Chen CX, Xu H. Self-assembly of surfactant-like peptides and their applications. Sci China Chem. 2014;57:1634–45.
Hollmann A, Martínez M, Noguera ME, Augusto MT, Disalvo A, Santos NC, Semorile L, Maffía PC. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide-membrane interactions of three related antimicrobial peptides. Colloid Surf B. 2016;141:528–36.
Jin L, Bai XW, Luan N, Yao HM, Zhang ZY, Liu WH, Chen Y, Yan XW, Rong MQ, Lai R, Lu MQ. A designed tryptophan- and lysine/arginine-rich antimicrobial peptide with therapeutic potential for clinical antibiotic-resistant Candida albicans Vaginitis. J Med Chem. 2016;5:1791–9.
Klok HA, Hwang JJ, Hartgerink JD, Stupp SI. Self-assembling biomaterials: l-lysine-dendron-substituted cholesteryl-(l-lactic acid)n. Macromolecules. 2015;35:6101–11.
Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. P Natl Acad Sci USA. 1988;85:5879–83.
Huang Z, Zhang C, Chen S, Ye F, Xing XH. Active inclusion bodies of acid phosphatase PhoC: aggregation induced by GFP fusion and activities modulated by linker flexibility. Microb Cell Fact. 2013;12:1–9.
Paudel MK, Sakamoto S, Van HL, Tanaka H, Miyamoto T, Morimoto S. The effect of varying the peptide linker length in a single chain variable fragment antibody against wogonin glucuronide. J Biotechnol. 2017;251:47–52.
Li J, Wang C, Hu D, Yuan F, Li X, Tang S, Wu M. Engineering a family 27 carbohydrate-binding module into an Aspergillus usamii β-mannanase to perfect its enzymatic properties. J Biosci Bioeng. 2017;123:294–9.
Pires DAT, Bemquerer MP, Nascimento CJD. Some mechanistic aspects on fmoc solid phase peptide synthesis. Int J Pept Res Ther. 2014;20:53–69.
Mihklepp K, Kivirand K, Nikopensius M, Nikopensius M, Peedel D, Utt M, Rinken T. Design and production of antibodies for the detection of Streptococcus uberis. Enzym Microb Tech. 2017;96:135–42.
Chen Q, Zheng J, Yuan X, Wang J, Zhang L. Folic acid grafted and tertiary amino based pH-responsive pentablock polymeric micelles for targeting anticancer drug delivery. Mater Sci Eng C Mater Biol Appl. 2017;82:1–9.
Chen Y, Yuan L, Zhou L, Zhang ZH, Cao W, Wu QQ. Effect of cell-penetrating peptide-coated nanostructured lipid carriers on the oral absorption of tripterine. Int J Nanomed. 2012;7:4581–91.
Makvandi P, Ghaemy M, Ghadiri AA, Mohseni M. Photocurable, antimicrobial quaternary ammonium-modified nanosilica. J Dent Res. 2015;94:1401–7.
Makvandi P, Ghaemy M, Mohseni M. Synthesis and characterization of photo-curable bis-quaternary ammonium dimethacrylate with antimicrobial activity for dental restoration materials. Eur Polym J. 2016;74:81–90.
Makvandi P, Corcione CE, Paladini F, Gallo AL, Montagna F, Jamaledin R, Pollini M, Maffezzoli A. Antimicrobial modified hydroxyapatite composite dental bite by stereolithography. Polym Advan Technol. 2017;29:364–71.
Zegers N, Gerritse K, Deen C, Boersma W, Claassen E. An improved conjugation method for controlled covalent coupling of synthetic peptides to proteins using glutaraldehyde in a dialysis method. J Immunol Methods. 1990;130:195–200.
Yang Y, Jiang JS, Du B, Gan ZF, Qian M, Zhang P. Preparation and properties of a novel drug delivery system with both magnetic and biomolecular targeting. J Mater Sci. 2009;20:301–7.
Li H, Jiang H, Zhao M, Fu Y, Sun X. Intracellular redox potential-responsive micelles based on polyethylenimine-cystamine-poly(ε-caprolactone) block copolymer for enhanced miR-34a delivery. Polym Chem. 2015;6:1952–60.
Lee SJ, Min KH, Lee HJ, Koo AN, Rim HP, Jeon BJ, Jeong SY, Heo JS, Lee SC. Ketal cross-linked poly(ethylene glycol)-poly(amino acid)s copolymer micelles for efficient intracellular delivery of doxorubicin. Biomacromolecules. 2011;12:1224–33.
Bengmark S. Nutrition of the critically ill - emphasis on liver and pancreas. Hepatobiliary Surg Nutr. 2012;1:25–52.
Dai Y, Li P, Wang A. Intelligent drug delivery system of intelligent high polymer materials. Prog Chem. 2007;19:362–9.
Reddy NH, Patnala S, Löbenberg R, Kanfer I. In vitro dissolution of generic immediate-release solid oral dosage forms containing BCS class I drugs: comparative assessment of metronidazole, zidovudine, and amoxicillin versus relevant comparator pharmaceutical products in South Africa and India. AAPS Pharm Sci Tech. 2014;15:1076–86.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) [grant number 31671797], Natural Science Foundation of Anhui University [grant number 2017KJA123, KJ2017A251 and KJ2018A0117], Anhui Natural Science Foundation [grant number 1808085QC85] and the Youth talent support program of Anhui Polytechnic University [grant number 2016BJRRC006].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ge, F., Qiao, Q., Zhu, L. et al. Preparation of a tumor-targeted drug-loading material, amphiphilic peptide P10, and analysis of its anti-tumor activity. J Mater Sci: Mater Med 30, 3 (2019). https://doi.org/10.1007/s10856-018-6204-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-018-6204-8