
Abstract
Multi-walled carbon nanotube (MWCNT)–Bioglass (BG) matrix composite was fabricated using a facile and scalable aqueous colloidal processing method without using any surfactants followed by spark plasma sintering (SPS) consolidation. The individual MWCNTs were initially uniformly dispersed in water and then entirely immobilized on the BG particles during the colloidal processing, avoiding their common re-agglomeration during the water-removal and drying step, which guaranteed their uniform dispersion within the dense BG matrix after the consolidation process. SPS was used as a fast sintering technique to minimise any damage to the MWCNT structure during the high-temperature consolidation process. The electrical conductivity of BG increased by 8 orders of magnitude with the addition of 6.35 wt% of MWCNTs compared to pure BG. Short-duration tests were used in the present study as a preliminary evaluation to understand the effect of incorporating MWCNTs on osteoblast-like cells. The analysed cell proliferation, viability and phenotype expression of MG-63 cells showed inhibition on 45S5 Bioglass®–MWCNT composite surfaces.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
There is an increasing need to develop novel materials for applications in tissue engineering. Bone scaffolds play a crucial role in bone regeneration by providing a support structure for bone defect sites and facilitating cell adhesion, proliferation and functionalities [1, 2]. Bone scaffolds should have enough bio-compatibility so that they can be absorbed in vivo by replacing new bone without disturbing the functions of the human body. Besides having good biological properties, bone scaffolds need to have sufficient mechanical properties to support hard tissue repair [3, 4]. 45S5 Bioglass® (BG) with osteoconductive, anti-bacterial, anti-inflammatory and potential angiogenic effects has been considered an excellent material for producing bone tissue scaffolds [5–10]. Bioglass, developed in 1971 by Prof. Larry Hench, has received increased attention due to its excellent properties in the field of biomaterials and regenerative medicine [11, 12]. Since the surfaces of biomaterials are in direct contact with the biological environment of the body, the surface properties of BG plays a crucial role in governing its biomedical applications. Hence, surface modification approaches are continuously being investigated for improving the biocompatibility and bioactivity of biomaterials [13].
The preparation of bioglass composites containing nanostructures has been a subject of growing interest in the recent scientific literature [14]. Especially carbon nano-materials with their unique combination of mechanical, electrical and thermal properties. They have found use in a wide range of applications including the biomedical sector [15–29]. Carbon nano-materials (graphene family) have been used to modify the functionality of biomaterials because of their unique ability to stimulate protein adhesion along with improved cellular functions [30]. The main advantages of using carbon nano-materials for biomedical applications include: (1) incorporation of nano-roughness on BG surface, which can lead to enhanced cell attachment and proliferation [31]; (2) improved electrical conductivity can help in cell stimulation by physioelectrical signal transfer with related beneficial effects on cell growth and tissue regeneration [32]; and (3) improved mechanical properties (i.e. strength and fracture toughness), which can strengthen the concept of BG scaffolds for biomedical applications [2, 33]. According to Porwal et al., the incorporation of graphene nanoplatelets (GNP) in BG resulted in an enhancement of the rate of hydroxyapatite formation (in vitro testing) compared to pure BG. This was ascribed to the nano surface roughness introduced by the GNP in the composite. They reported an increase of 9 orders of magnitude in the electrical conductivity of BG with 5 vol % loading of GNP [34]. Similarly Meng et al. produced CNT coated BG scaffolds by the foam replication method and observed no significant difference in the cell culture of mesenchymal stem cells (MSCs) for coated and uncoated CNT scaffolds [35]. From a structural point of view, Jia et al. reported an improvement in the flexural strength (159 %) and fracture toughness (105 %) of 45S5 BG reinforced with CNTs [36]. In the presence of electrical simulation in the case of polycarbonate urethane (PCU)-CNT film, Khang et al. reported an enhancement of up to 50 % in chondrocyte adhesion and long term cell densities (up to 2 days) as compared to the pure PCU. With electrical simulation, there was a more than 200 % improvement in cell density, confirming the positive effect of the enhanced electrical conductivity of the PCU-CNT film [32].
Although CNTs possess various advantageous properties for tissue engineering, there are concerns about their cytotoxicity and biodegradation [37, 38]. The reaction of the cells to the CNTs depends on three major factors: (1) purity; (2) morphology and; (3) loading of the nano-filler. Purity of the carbon nano-fillers is very important. CNTs contain metal nano-particles as impurities which reduces the bioactivity of CNTs. Another problem is the non-solubility of CNTs in water based solvents, which means that non-biocompatible solvents need to be used for processing. Acid treatment of CNTs can solve both of these problems; it removes metal nano-particles and functionalizes the CNTs, making them water soluble. Authors have reported improvement in bio-activity with functionalisation of CNTs [39]. The morphology of carbon nano-materials also plays a crucial role in understanding the bioactivity of such materials. Since CNTs have fibrous needle like shape, their shape might affect the bioactivity of the prepared composites. Authors have reported that CNTs damage the lysosomal membranes in cells [37, 40]. Also, the loading of CNTs in bio-materials can play a crucial role in determining its bioactivity. The bioactivity of nano-composites mainly depends on the interaction of the cells with the incorporated nano-phase since the matrix is already bioactive. Although, high loading of CNTs can have various advantages, like improved mechanical and electrical properties, their effect in cell environments are still not clear. So far, no extensive report has been published on the material processing and the in vitro cell culture behaviour of the BG–MWCNT composites. In our work we report the fabrication of well dispersed MWCNT–BG matrix composite using a facile and scalable aqueous colloidal processing method without using any surfactants followed by spark plasma sintering (SPS) consolidation. The details about the processing and characterizations of the BG and BG–MWCNT composite are provided in the following sections. We clarify the effects of MWCNTs incorporation on the in vitro cell culture response of BG. Therefore, human osteoblast-like cells were used to evaluate their cell morphology cultured on the different BG samples.
2 Materials and methods
In order to prepare well dispersed MWCNT (6.35 wt%)-BG matrix composite powder, a facile and scalable aqueous colloidal processing method was used. The processing steps were developed to: (1) achieve good dispersion of individual MWCNTs within the BG matrix without using any potentially toxic surfactants; and (2) sinter the composite without damaging the MWCNTs structure. A loading of 6.35 wt% MWCNTs was selected because previous work showed that this produced optimum mechanical properties [36]. This loading is also sufficient to form a percolating network of MWCNTs to make the BG matrix electrically conductive. Details about the processing and characterisations are provided in the following sections.
2.1 Preparation of a highly stable, aqueous MWCNT colloidal suspension
Highly pure pristine MWCNTs (Hodogaya Chemical Co., Ltd.) synthesized by a catalytic chemical vapour deposition process and graphitized/purified at approximately 3000 °C were used as the starting material. To make them dispersible and colloidally stable in water, their hydrophobic surfaces were functionalized using a controlled acid treatment process. The details are described elsewhere [41–47]. The resulting functionalized MWCNTs were hydrophilic and still highly crystalline [41–44, 47]. Briefly 1.4 g of the pristine MWCNTs was refluxed in 120 mL of 3:1 v/v ratio of 98 % H2SO4 and 68 % HNO3 acid mixture for 20 min at 110 °C. The treated MWCNTs were then thoroughly washed with ultrapure water and filtered using a 1.2 μm membrane, probe sonicated, and dispersed in ultrapure water (2.8 g/L) for 10 h while being cooled continuously. The natural pH of the as-prepared highly stable suspension was ~4.1 with a zeta potential value of ~−40 mV. The pH of the 10 h sonicated MWCNT suspension was then adjusted to ~11.7 (zeta potential ~−55 mV) using NaOH solution, before being added to the aqueous BG dispersion.
2.2 Preparation of composite powder
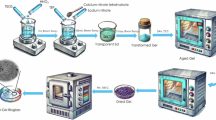
The steps for the preparation of the powder mixture are illustrated in Fig. 1. Briefly, 20.6 g of BG powder was dispersed in 550 ml of ultrapure water (~37 g/L) and probe-sonicated for 3 h while being vigorous magnetically stirred. This process was performed to break the particles aggregates so that they would provide a better host for the MWCNTs. The natural pH and zeta potential of the resulting dispersion were ~11.7 and ~−32 mV respectively. The BG dispersion, however, had poor colloidal stability mainly due to the large micro-meter scale size of the particles. Therefore, combined sonication and stirring was performed to prevent the BG particles agglomeration prior to the addition of the MWCNTs.

Schematic showing processing steps involved in the fabrication of well dispersed MWCNT–BG matrix composite
The MWCNTs aqueous suspension was then rapidly added to the vigorously stirred and sonicated BG dispersion and the mixture was vigorously stirred and sonicated for 10 min to allow the uniform dispersion of the MWCNTs with the particles. The mixture was then vigorously stirred for 2 h (no sonication) before being left stagnant. The stirring process could have enabled numerous collisions between the MWCNTs and BG particles leading to the generation of attractive van der Waals forces that could overcome the repulsive electrostatic forces, thus resulting in the formation of MWCNT-coated BG particles. After 10 min of stagnation of the mixture, all of the MWCNTs and BG particles were settled leaving behind a transparent, MWCNT-free supernatant, this suggests the arrest and immobilization of the entire MWCNTs by the BG particles. The settled powder (~6.35 wt% MWCNTs) was collected and dried overnight in air at 100 °C followed by grinding and sieving of the powder using a 250 mesh sieve.
2.3 Sintering and characterizations
Bioglass powder (as received) and BG–MWCNT (6.35 wt%) composite powders were sintered into 1 cm disks of 3 mm thickness using an SPS furnace (HPD 25/1, FCT systems, Germany) at 550 °C with a 2 min dwell time, 70 MPa pressure and in a vacuum environment [34, 48, 49]. The temperature was raised at a heating rate of 50 °C/min up to 500 °C and then 10 °C/min from 500 to 550 °C/min to avoid overheating of the BG and BG–MWCNT composite. It should be noted that BG and BG–MWCNT powders were held in the SPS furnace at 300 °C for 5 min to remove any trapped solvent from the powders after processing. Pressure was applied from the start and kept constant throughout the sintering process. The sintered samples (10 mm diameter, 3 mm thickness discs) were then grounded and polished down to 4000 grit using SiC paper. The theoretical densities of the composite were estimated using the rule of mixture and taking the densities of BG and MWCNTs to be 2.72 and 2.1 g/cm3, respectively, while bulk densities were calculated using Archimedes’ method [44]. Raman spectroscopy (Renishaw Ramascope, He-Neon Laser-633 nm) was performed on the sintered samples in order to analyse any damage to the MWCNTs during high temperature processing. The dense samples were characterized using SEM (JEOL JSM-6300). Since, BG–MWCNT composite was electrically conductive there was no need to have a gold/carbon coating on the sample. Samples were fractured and the fractured surfaces were analysed directly in the SEM. XRD (Siemens Diffraktometer-D5000) was done in reflection mode with Cu-Kα incident radiation from 5 to 70° (2 theta) and step size of 0.033o. The electrical conductivity measurements were performed using a two point probe method. The voltage was applied from 0 to 1 V using an Agilent voltmeter in steps of 0.1 V and I–V curves were recorded. The resistivities of the samples were determined using the formula ρ = (R × A)/L, where R is the resistance, A is the cross sectional area and L is the length of the sample. Silver paste was used to prepare the electrodes on the cross sectional areas of the samples.
2.4 In vitro biocompatibility studies
2.4.1 Cell culture and analysis
Since the morphological organization of osteoblasts exhibits cell-substrate adhesion, it would be a sensitive indicator of the biocompatibility of a biomaterial. To evaluate the cell compatibility of the MWCNT-modified BG compared with the standard BG, a osteoblast-like cell line, MG-63 (Sigma), was used (Fig. 2). MG-63 cells express mainly elongated and polygonal phenotype with many filopodias. These cells were cultured at 37 °C in an atmosphere of 95 % humidified air and 5 % CO2, in DMEM (Dulbecco, Germany) containing 10 vol% fetal bovine serum (FBS, Sigma-Aldrich, Germany) and 1 vol% penicillin/streptomycin (Life technology, Germany). Cells were grown for 48 h to confluence in 75 cm2 culture flasks (Nunc, Denmark), before being harvested using Trypsin/EDTA (Sigma, Germany). They were counted by a hemocytometer (Roth, Germany) and diluted to a final concentration of 1 × 105 cells/ml.

Light microscope image of the applied MG-63 cell line
Before cell seeding, the samples were sterilized by dry heating at 160 °C for 7 h. To reduce the well-known pH shift of the BG, samples were pre-cultivated in a culture media for 3 days. Afterwards the cells were seeded in direct contact in a 48-well cell culture plate (Greiner, Germany). To compare the morphology of the cell grown on the test samples, cell culture dishes were used as a reference material.
Cell proliferation was evaluated using the BrdU labelling and detection kit (Roche, Germany). Briefly, after 48 h of incubation, the samples were removed from the culture medium, immersed in 10 μM BrdU labeling medium and placed in an incubator (5 % CO2, 37 °C). After 2 h the BrdU labeling medium was aspirated and the cells were fixed by addition of 1 ml FixDenat and incubated at 25 °C. After 30 min, the fixing solution was aspirated and 1 ml of anti-BrdU-POD working solution was added. The samples were again incubated at 25 °C. After 90 min, the antibody conjugate was aspirated and the scaffolds were washed three times with washing buffer solution. After drying, the samples were incubated with 1 ml of substrate solution at 25 °C until the color development was sufficient for photometric detection. The reaction was stopped by the addition of 250 μl of 1 M H2SO4 and the absorbance of the samples was measured in an ELISA reader (PHOmo, anthos Mikrosysteme GmbH, Germany) at 450 nm.
The water soluble tetrasodium (WST) assay was used to determine cell viability. After cell cultivation, the cell culture medium was removed and samples were washed with 0.5 mL phosphate buffered saline (PBS). Afterwards, 0.25 mL WST medium (containing 1 vol% of WST reagent (Cell Counting Kit-8, Sigma) and 99 vol% of DMEM medium) was added and incubated for 2 h. After incubation, 0.1 mL of the supernatant was transferred to a 96-well culture plate and spectrometically measured using a microplate reader (PHOmo, anthos Mikrosysteme GmbH, Germany) at 450 nm.
2.4.2 Cell visualization
To visualize the cells attachment and distribution on the samples, the cell cultures were evaluated using fluorescence and scanning electron microscopy, which were performed as follow:
2.4.2.1 Fluorescence microscopy (FM)
To observe the formation of cytoskeleton and cell distribution in contact with BG samples, fluorescence microscopy (FM, Scope.A1, Carl Zeiss) was used after staining. After the cultivation period of 48 h, the adherent cells were fixed with 3.7 vol% paraformaldehyde for 10 min and permeabilised with 0.1 vol% Triton X-100 (in PBS) for 10 min at room temperature. Green fluorescent Sytox (Molecular Probes) and red fluorescent Rhodamine Phalloidin (Molecular Probes®) were used for cell staining. To detect the cytoskeleton, the cells were incubated for 60 min with phalloidin (diluted 1:50 by volume) at room temperature followed by incubation with 1 µg/ml Sytox for 5 min. Samples were washed and left in PBS for microscopic imaging.
2.4.2.2 Scanning electron microscopy (SEM)
The cell cultures were washed with PBS, fixed with a solution containing 3 vol% glutaraldehyde (Sigma, Germany) and 3 vol% paraformaldehyde (Sigma, Germany) in 0.2 M sodium cacodylate buffer (pH 7.4), and finally rinsed with PBS for SEM analysis (Auriga CrossBeam, Carl Zeiss Microscopy GmbH, Germany). All samples were dehydrated in graded ethanol series (30, 50, 70, 80, 90, 95, and 99.8 vol%). Samples were maintained at 99.8 vol% ethanol and critical-point dried (EM CPD300, Leica, Germany).
2.4.3 Statistics
Results are presented using the mean value and standard deviation of four replicates of each sample type. All results were normalized to cell growth on a Petri dish (REF = 100 %). The differences in analysis parameters between the different samples investigated were evaluated by one-way analysis of variance (ANOVA). The level of the statistical significance was defined at P < 0.05 (Origin 8.1G, Origin Lab Corporations, USA). The significance level was set as *P < 0.05, **P < 0.01 and ***P < 0.001. For the comparison of the mean values the Tukey test was used.
3 Results and discussion
3.1 Sintering, microstructural characterisation and electrical conductivity
Figure 3a shows the SPS data during sintering of BG and BG–MWCNT composite. Speed (piston travel rate, black curve), relative piston travel (blue curve) and temperature (red curve) were plotted to evaluate any changes in the sintering curve of BG with the addition of MWCNTs. The sintering curve of pure BG showed a single sharp peak at 490 °C suggesting viscous flow as the sintering mechanism. In comparison, the sintering curve of the BG–MWCNTs composite became broader with a sintering peak at 510 °C. The addition of MWCNTs to the BG matrix increased the viscosity of the BG–MWCNT composite, which leads to an increase in the sintering temperature and broadening of the sintering peak. Similar behaviour was observed for BG-graphene nanoplatelet (GNP) composites, in which case the sintering temperature was increased by 50 °C with the addition of GNPs [34]. Figure 3b shows the XRD spectra of BG and BG–MWCNT composite. XRD confirmed the crystallisation of the BG and BG–MWCNT composite after sintering at 550 °C. Na2CaSi3O8 (PDF 00-012-0671) was observed as the main crystalline phase for both BG and BG–MWCNT composite. In the case of BG–MWCNT, there was an overlapping of the peaks at 26.3o of the Na2CaSi3O8 phase and MWCNTs. Comparing the XRD spectra of both materials it was clear that BG was more crystallised compared to BG–MWCNT composite. MWCNTs increased the viscosity of the BG matrix, and thus retarded the densification (Fig. 3a) and inhibited the crystallisation. A similar behaviour has been reported for other CNT and GNP composites [34, 50].

a Sintering profiles showing speed (piston travel rate), relative piston travel, temperature; and b XRD spectra of BG and BG–MWCNT (6.35 wt%) composite sintered at 550 °C with 2 min dwell time and 70 MPa pressure
In order to confirm the damage to MWCNTs during high temperature processing Raman spectroscopy was performed. Figure 4 shows the Raman spectra of BG–MWCNTs powders (before SPS) and BG–MWCNTs bulk sample (after SPS). Typical D, G and D′ peaks corresponding to MWCNTs were observed for both BG–MWCNTs powders and bulk samples. The D band in MWCNTs corresponds to disorder and defects, the G band represents the in plane vibrations of the Carbon–Carbon bonds and D’ band is attributed to the overtone of the D band [51]. The ratio of the intensities of D and G bands (ID/IG) can be used to quantify the damage induced in composites during high temperature processing. The ID/IG ratio in the case of BG–MWCNTs composite powders was found to be 0.54 which increased to 0.97 after SPS processing suggesting some damage was induced to MWCNTs during high temperature processing. Also D, G and D’ peaks in the case of BG–MWCNTs bulk samples (after SPS) were shifted to higher wavenumbers by 7, 12 and 13 cm−1 respectively when compared to BG–MWCNTs powders (before SPS). The up shift in D, G and 2D bands in the case of BG–MWCNTs bulk samples (after SPS) was produced by compressive residual stresses introduced on MWCNTs due to mismatch in elastic moduli and thermal coefficients of expansions of BG and MWCNTs [41, 46, 51]. Similar behaviour has also been reported for BG-graphene nano-platelet composites [34].

Raman spectra of BG–MWCNTs composite powders (before SPS) and bulk samples (after SPS)
Table 1 shows the bulk and relative theoretical densities, and electrical conductivities of the BG and BG–MWCNT (6.35 wt%) composite. The density measurements confirmed ~99 % relative density for the BG–MWCNT composite compared to ~100 % density in the case of pure BG. This could be related to the increased viscosity of the BG–MWCNT composite, which was also observed in its sintering profile. The electrical conductivity of BG–MWCNT composite increased by 8 orders of magnitudes compared to pure BG. The increase in electrical conductivity was due to the formation of a percolating network of MWCNTs in the BG matrix. The formation of a percolating network was also confirmed by SEM images. Figure 5 shows SEM images of a fracture surface of BG–MWCNT composite, which shows the distribution of individual MWCNTs within the dense BG matrix. MWCNTs were found to be uniformly and individually distributed throughout the BG matrix. The BG particles immobilized the entire well-dispersed MWCNTs in water during the colloidal processing, which avoided the common re-agglomeration of MWCNTs during the water-removal and drying step, and resulted in a uniform distribution of MWCNTs within the dense BG matrix after the SPS consolidation. The good interface between BG and MWCNTs was confirmed by analysing the high magnification SEM images of the fractured surface of the composite. Figure 5 (c) shows exposed MWCNTs on the fracture surface, which were either pulled out from the BG matrix or failed/buckled during the composite fracture [46, 52]. Since the interface between BG and MWCNTs was strong in shear and due to presence of residual compressive stresses on the MWCNTs in the radial direction (Fig. 4), the MWCNTs underwent a unique multiwall-type failure resulting in more energy dissipation during the fracture [47, 52]. MWCNTs failure occurred mainly due to strong interfacial shear resistance which would not be possible without an intimate interface between BG and MWCNTs [44, 47]. It should be noted that MWCNTs used in the present study were dispersed in the BG matrix using water as the solvent without the use of any potentially toxic surfactants. This novel non-toxic and environmentally friendly approach can be used for the fabrication of composites containing nanostructure reinforcements (1D or 2D).

a Low; and b high magnification SEM images of fracture surfaces of BG–MWCNT (6.35 wt%) composite showing the uniform distribution of individual MWCNTs within the dense BG matrix; c shows the exposed MWCNTs on the fracture surface, which were either pulled out from the BG matrix or failed/buckled during the composite fracture due to strong interfacial shear resistance
3.2 In vitro biocompatibility
Due to their excellent mechanical properties, CNT (both multiwall and single wall CNT) are of high interest in the preparation of reinforced biodegradable composites for biomedical applications where mechanically resistant but still bioactive materials are required [37, 38, 40, 53]. Although the effect of CNT on the cellular metabolism in bone cells still remains unclear, CNT have been considered to impart a beneficial role on bone cell behaviour [31, 32, 35]. In vitro tests with established cell lines (e.g. MG-63) are usually carried out for 24–48 h when a completely new biomaterial is tested in order to generate reproducible results about the interactions between cells and the biomaterial [54–58]. Such test setups are created to determine eventual changes in the cell proliferation behaviour as well as to assess modifications in cell morphology caused by the material. Such short-duration tests were used in this study as a preliminary evaluation to understand the effect of incorporating MWCNTs on MG63 cells. In Fig. 6 cell proliferation measured by DNA-synthesis and cell viability detected by mitochondrial activity of MG-63 cells is shown after 48 h of incubation. Both materials induced a considerably reduction of cell activities compared to the REF material. Furthermore, cell proliferation of MG-63 cells was significantly (P < 0.001) inhibited on the MWCNT samples. Similar results were obtained using the cell viability measurement.

Cell proliferation and cell viability of MG-63 osteoblast-like cells on BG, BG–MWCNT composite (6.35 wt%) and REF after 48 h of cultivation
Figure 7 shows representative fluorescence microscopy images of MG-63 cells on different surfaces after 48 h of cultivation. In the case of the BG sample, the surface seems to be covered completely by MG-63 cells. Compared to the cell culture dish (REF), MG-63 cells expressed on BG have the typical osteoblastic phenotype: mainly elongated and polygonal phenotype with many filopodias and well-formed strongly expressed actin fibers. For the BG–MWCNT composite, the osteoblasts were less attached; exhibiting less spreading behavior, a much smaller diameter and a more cuboidal shape. There were few or even no cytoskeleton formed in the MG-63 cells. After the initial adhesion, MG-63 cells developed their cytoskeleton spreading on the biomaterial surface. For MG-63 cells thin actin fibers can be detected around the nuclei. On the REF surface, osteoblasts showed similar behavior as seen for cells on BG. They were well spread with a distinctive, filamentous actin network.

Nucleus and actin formation of MG-63 osteoblast-like cells on; a BG; b BG–MWCNT composite (6.35 wt%); and c REF after 48 h of cultivation
After the initial adhesion, MG-63 develops their cytoskeleton spreading on the BG and REF surfaces. However, it seems that BG–MWCNT composite inhibited the adhesion of cells. The MG-63 cells are seen to be rounded. Interestingly, comparing MG-63 and rat mesenchymal stem cells cultured on BG for 48 h, as investigated previously [59], the MG-63 cells exhibited a more rounded morphology with pseudopodia extending across peaks. In general, although the samples with MWCNT exhibited some degree of toxicity, different cell types (osteoblasts, fibroblasts, endothelial cells, etc.) respond to biomaterial surfaces in different manners.
The in vitro and in vivo biocompatibility of MWCNTs is controversially discussed in the literature [37, 38, 40]. For example, Ghosh et al. reported genotoxic effects of MWCNTs on bone marrow stromal cells [60]. The detected DNA fragmentation shown in the Ghosh et al. study could also be the reason for the reduced DNA-synthesis in MG-63 cells found here (Fig. 6). As mentioned above, it was shown that lysosomal damage is one of the main reasons for CNTs leading to apoptotic and necrotic cell death [40]. This effect could be the reason for the inhibition of proliferation in MG-63 cells cultured on MWCNT containing samples for 48 h. Furthermore, the measured reduced mitochondrial activity could be caused by direct injury from the MWCNT or indirectly by fragments of the damaged lysosome [40]. Nevertheless, it is interesting to put the present results in the context of the available literature on CNT-bioactive glass composites. In a relevant study involving CNT coated 45S5 BG-based scaffolds, Meng et al. [35] also showed an inhibition of viability of MSC on CNT coated scaffolds compared to pure BG scaffolds during 4 weeks of incubation. Simultaneously, however, an increase of the measured ALP was measured suggesting improved in vitro osteogenesis imparted by the MWCNT coating. In another study, MG-63 cells cultured on poly(3-hydroxybutyrate) (P(3HB)) composites with bioactive glass particles and MWCNTs exhibited a reduced cell behaviour after 24 h of contact with samples containing MWCNT and the authors stated that MWCNTs as fillers in a polymeric matrix at concentrations >2 wt % could have an inhibition influence on cell proliferation [61]. Moreover, polysulfone films covered with and without MWCNTs were also tested with fibroblasts and osteoblasts for 7 days of incubation [62].
The measured cell viabilities, Il-6, collagen and osteocalcin synthesis indicated no negative cellular behaviour of the CNT. Generally, BG is a biocompatible and bioactive biomaterial exhibiting high surface reactivity. The cell morphology of MG-63 cells was observed by SEM after 48 h of incubation on the different sample surfaces (Fig. 8). MG-63 osteoblast-like cells spread extensively and were flattened on the BG surface. They were of polygonal shape with filopodia or very thin extensions. It seemed that the cells did not have any preferred orientation. Some of them appeared thicker in the central area of the nucleus and flattened in the peripheral regions. Furthermore, in some areas of the sample the MG-63 started to form a multilayer structure, although cell proliferation and viability of MG-63 cells on BG is strongly reduced compared to the reference material after 48 h of cultivation (Fig. 6). The cell density on BG–MWCNT composite was much lower compared to pure BG samples. In addition, the morphology of MG-63 osteoblast-like cells on BG–MWCNT was polygonal and some cells showed a delayed spreading. As discussed previously, bioactivity of nano-materials depend on various factors including purity, morphology and loading of CNTs. Consequently, if cell DNA is fragmented or cell organelles like lysosomes are damaged through MWCNT, the cell morphology is also affected by the event. Cell spreading and cytoskeleton expression are inhibited through apoptotic and necrotic effects. Several studies could demonstrate that changes in the cell morphology are typical effect of CNTs on adherent growing cells [63, 64]. Although there has been extensive research on CNT reinforced biomaterials, our understanding of the cytotoxicity of CNTs is still inconclusive and more research is needed on this topic to gain further knowledge on the mechanism of interaction of CNT (embedded in a matrix material or protruding from a surface) with relevant cells.

SEM images of cell morphology of MG-63 osteoblast-like cells on; a BG; b BG–MWCNTs composite (6.35 wt%); and c REF after 48 h of cultivation
4 Conclusions
Well dispersed MWCNT–BG matrix composite powder was prepared using a facile and scalable aqueous colloidal processing route without using any surfactants. BG and BG–MWCNT powders were sintered using SPS at 550 °C with 2 min dwell time under 70 MPa pressure. MWCNTs were shown to be individually and uniformly distributed within the dense BG matrix, which is due to their entire immobilization on the BG particles during the colloidal processing step, avoiding their common re-agglomeration during the water-removal and drying step. The electrical conductivity of BG increased by 8 orders of magnitude with the addition of 6.35 wt% MWCNTs, which is due to the formation of a percolating network of MWCNTs within the dense BG matrix. The in vitro cell activity results on the materials evaluated using a cell line (MG-63 osteoblast-like) suggests that BG–MWCNT composite inhibited the attachment of cell line cultures. Fewer cells were observed on the composite surface. The cytoskeleton was not expressed and no dense monolayer was formed for BG–MWCNT composite compared to pure BG. For that reason, further studies on BG–MWCNT composite substrates should be carried out for longer cell culture times to assess the long-term biological compatibility of the material.
References
Hoppe A, Guldal NS, Boccaccini AR. A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials. 2011;32(11):2757–74. doi:10.1016/j.biomaterials.2011.01.004.
Shuai CJ, Gao CD, Feng P, Peng SP. Graphene-reinforced mechanical properties of calcium silicate scaffolds by laser sintering. Rsc Adv. 2014;4(25):12782–8. doi:10.1039/C3ra47862a.
Woodruff MA, Lange C, Reichert J, Berner A, Chen FL, Fratzl P, et al. Bone tissue engineering: from bench to bedside. Mater Today. 2012;15(10):430–5.
Jones JR. New trends in bioactive scaffolds: The importance of nanostructure. J Eur Ceram Soc. 2009;29(7):1275–81. doi:10.1016/j.jeurceramsoc.2008.08.003.
Hench LL. Bioceramics—from Concept to Clinic. J Am Ceram Soc. 1991;74(7):1487–510. doi:10.1111/j.1151-2916.1991.tb07132.x.
Chen QZ, Efthymiou A, Salih V, Boccaccini AR. Bioglass®-derived glass–ceramic scaffolds: study of cell proliferation and scaffold degradation in vitro. J Biomed Mater Res A. 2008;84A(4):1049–60. doi:10.1002/jbm.a.31512.
Xynos ID, Edgar AJ, Buttery LDK, Hench LL, Polak JM. Ionic products of bioactive glass dissolution increase proliferation of human osteoblasts and induce insulin-like growth factor II mRNA expression and protein synthesis. Biochem Bioph Res Co. 2000;276(2):461–5. doi:10.1006/bbrc.2000.3503.
Day RM, Boccaccini AR, Shurey S, Roether JA, Forbes A, Hench LL, et al. Assessment of polyglycolic acid mesh and bioactive glass for soft-tissue engineering scaffolds. Biomaterials. 2004;25(27):5857–66. doi:10.1016/j.biomaterials.2004.01.043.
Zhang D, Lepparanta O, Munukka E, Ylanen H, Viljanen MK, Eerola E, et al. Antibacterial effects and dissolution behavior of six bioactive glasses. J Biomed Mater Res A. 2010;93A(2):475–83. doi:10.1002/Jbm.A.32564.
Gorustovich AA, Roether JA, Boccaccini AR. Effect of bioactive glasses on angiogenesis: a review of in vitro and in vivo evidences. Tissue Eng Part B-Rev. 2010;16(2):199–207. doi:10.1089/ten.teb.2009.0416.
Hench LL, Splinter RJ, Allen WC, Greenlee TK. Bonding mechanisms at the interface of ceramic prosthetic materials. J Biomed Mater Res A. 1971;5(6):117.
Hench LL. The story of Bioglass (R). J Mater Sci-Mater M. 2006;17(11):967–78. doi:10.1007/s10856-006-0432-z.
Anderson AB, Dallmier AW, Chudzik SJ, Duran LW, Guire PE, Hergenrother RW, et al. Technologies for the surface modification of biomaterials. In: Yaszemski MJ, Trantolo DJ, Lewandrowski K-U, Hasirci V, Altobelli DE, Wise DL, editors. Biomaterials in Orthopedics. 2nd ed. CRC Press; 2003. p. 93–148.
Wang JX, Fan YB, Gao Y, Hu QH, Wang TC. TiO2 nanoparticles translocation and potential toxicological effect in rats after intraarticular injection. Biomaterials. 2009;30(27):4590–600. doi:10.1016/j.biomaterials.2009.05.008.
Geim AK, Novoselov KS. The rise of graphene. Nat Mater. 2007;6(3):183–91.
Lee C, Wei XD, Kysar JW, Hone J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science. 2008;321(5887):385–8. doi:10.1126/science.1157996.
Balandin AA, Ghosh S, Bao WZ, Calizo I, Teweldebrhan D, Miao F, et al. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008;8(3):902–7. doi:10.1021/Nl0731872.
Pumera M. Electrochemistry of graphene: new horizons for sensing and energy storage. Chem Rec. 2009;9(4):211–23. doi:10.1002/tcr.200900008.
Porwal H, Grasso S, Reece M. Review of graphene-ceramic matrix composites. Adv Appl Ceram. 2013;112(8):443. doi:10.1179/174367613X13764308970581.
Mohanty N, Berry V. Graphene-based single-bacterium resolution biodevice and DNA transistor: interfacing graphene derivatives with nanoscale and microscale biocomponents. Nano Lett. 2008;8(12):4469–76. doi:10.1021/Nl802412n.
Khan U, May P, Porwal H, Nawaz K, Coleman JN. Improved adhesive strength and toughness of polyvinyl acetate glue on addition of small quantities of graphene. Acs Appl Mater Interface. 2013;5(4):1423–8. doi:10.1021/Am302864f.
Wu JB, Agrawal M, Becerril HA, Bao ZN, Liu ZF, Chen YS, et al. Organic light-emitting diodes on solution-processed graphene transparent electrodes. ACS Nano. 2010;4(1):43–8. doi:10.1021/Nn900728d.
Iijima S. Helical microtubules of graphitic carbon. Nature. 1991;354(6348):56–8. doi:10.1038/354056a0.
Inam F, Yan HX, Jayaseelan DD, Peijs T, Reece MJ. Electrically conductive alumina-carbon nanocomposites prepared by Spark Plasma Sintering. J Eur Ceram Soc. 2010;30(2):153–7. doi:10.1016/j.jeurceramsoc.2009.05.045.
Porwal H, Tatarko P, Grasso S, Khaliq J, Dlouhý I, Reece M. Graphene reinforced alumina nano-composites. Carbon. 2013;64:359–69. doi:10.1016/j.carbon.2013.07.086.
Porwal H, Tatarko P, Saggar R, Grasso S, Kumar Mani M, Dlouhý I et al. Tribological properties of silica–graphene nano-platelet composites. Ceram Int. 2014(0). doi:10.1016/j.ceramint.2014.04.046.
Porwal H, Grasso S, Mani MK, Reece MJ. In situ reduction of graphene oxide nanoplatelet during spark plasma sintering of a silica matrix composite. J Eur Ceram Soc. 2014(0). doi:10.1016/j.jeurceramsoc.2014.04.031.
White AA, Kinloch IA, Windle AH, Best SM. Optimization of the sintering atmosphere for high-density hydroxyapatite-carbon nanotube composites. J R Soc Interface. 2010;7:S529–39. doi:10.1098/rsif.2010.0117.focus.
White AA, Windle AH, Kinloch IA, Best SM. Preparation and properties of carbon nanotube-reinforced hydroxyapatite. Bioceramics. 2008;361–363:419–22. (Vol 20, Pts 1 and 2.).
Sanchez VC, Jachak A, Hurt RH, Kane AB. Biological interactions of graphene-family nanomaterials: an interdisciplinary review. Chem Res Toxicol. 2012;25(1):15–34. doi:10.1021/Tx200339h.
Lobo AO, Corat MAF, Antunes EF, Palma MBS, Pacheco-Soares C, Garcia EE, et al. An evaluation of cell proliferation and adhesion on vertically-aligned multi-walled carbon nanotube films. Carbon. 2010;48(1):245–54. doi:10.1016/j.carbon.2009.09.012.
Khang D, Park GE, Webster TJ. Enhanced chondrocyte densities on carbon nanotube composites: the combined role of nanosurface roughness and electrical stimulation. J Biomed Mater Res A. 2008;86A(1):253–60. doi:10.1002/Jbm.A.31803.
Porwal H, Tatarko P, Grasso S, Hu C, Boccaccini AR, Dlouhý I et al. Toughened and machinable glass matrix composites reinforced with graphene and graphene-oxide nano platelets. Sci Technol Adv Mat. 2013;14. doi:10.1088/1468-6996/14/5/055007.
Porwal H, Grasso S, Cordero-Arias L, Li C, Boccaccini A, Reece M. Processing and bioactivity of 45S5 Bioglass®-graphene nanoplatelets composites. J Mater Sci: Mater Med. 2014;1–11. doi:10.1007/s10856-014-5172-x.
Meng D, Rath SN, Mordan N, Salih V, Kneser U, Boccaccini AR. In vitro evaluation of 45S5 Bioglass®-derived glass-ceramic scaffolds coated with carbon nanotubes. J Biomed Mater Res Part A. 2011;99A:435–44.
Jia ZZ, Zhang J, Jia CC, Nie JH, Chu K. Preparation and characterization of mechanical properties of carbon nanotube/45S5Bioglass composites for biologic applications. Mat Sci Eng A-Struct. 2011;528(3):1553–7. doi:10.1016/j.msea.2010.10.077.
Madani SY, Mandel A, Seifalian AM. A concise review of carbon nanotube’s toxicology. Nano Rev. 2013;4. doi:10.3402/nano.v4i0.21521.
Li XM, Fan YB, Watari F. Current investigations into carbon nanotubes for biomedical application. Biomed Mater. 2010;5(2). doi:10.1088/1748-6041/5/2/022001.
Pichardo S, Gutierrez-Praena D, Puerto M, Sanchez E, Grilo A, Carnean AM, et al. Oxidative stress responses to carboxylic acid functionalized single wall carbon nanotubes on the human intestinal cell line Caco-2. Toxicol In Vitro. 2012;26(5):672–7. doi:10.1016/j.tiv.2012.03.007.
Andon FT, Fadeel B. Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc Chem Res. 2013;46(3):733–42. doi:10.1021/Ar300020b.
Estili M, Kawasaki A, Sakka Y. Highly concentrated 3D macrostructure of individual carbon nanotubes in a ceramic environment. Adv Mater. 2012;24(31):4322–6. doi:10.1002/adma.201201134.
Estili M, Kawasaki A. An approach to mass-producing individually alumina-decorated multi-walled carbon nanotubes with optimized and controlled compositions. Scripta Mater. 2008;58(10):906–9. doi:10.1016/j.scriptamat.2008.01.016.
Estili M, Kawasaki A, Sakamoto H, Mekuchi Y, Kuno M, Tsukada T. The homogeneous dispersion of surfactantless, slightly disordered, crystalline, multiwalled carbon nanotubes in alpha-alumina ceramics for structural reinforcement. Acta Mater. 2008;56(15):4070–9. doi:10.1016/j.actamat.2008.04.029.
Estili M, Sakka Y, Kawasaki A. Unprecedented simultaneous enhancement in strain tolerance, toughness and strength of Al2O3 ceramic by multiwall-type failure of a high loading of carbon nanotubes. Nanotechnology. 2013;24(15):155702.
Ramón-Azcón J, Ahadian S, Estili M, Liang X, Ostrovidov S, Kaji H, et al. Dielectrophoretically aligned carbon nanotubes to control electrical and mechanical properties of hydrogels to fabricate contractile muscle myofibers. Adv Mater. 2013;25(29):4028–34. doi:10.1002/adma.201301300.
Estili M, Sakka Y. Recent advances in understanding the reinforcing ability and mechanism of carbon nanotubes in ceramic matrix composites. Sci Technol Adv Mat. 2014;15(6). doi:10.1088/1468-6996/15/6/064902.
Estili M, Kawasaki A. Engineering strong intergraphene shear resistance in multi-walled carbon nanotubes and dramatic tensile improvements. Adv Mater. 2010;22(5):607–10. doi:10.1002/adma.200902140.
Grasso S, Sakka Y, Maizza G. Electric current activated/assisted sintering (ECAS): a review of patents 1906–2008. Sci Technol Adv Mat. 2009;10(5). doi:10.1088/1468-6996/10/5/053001.
Grasso S, Chinnam RK, Porwal H, Boccaccini AR, Reece MJ. Low temperature spark plasma sintering of 45S5 Bioglass®. J Non-Cryst Solids. 2013;362:25–9.
Cho J, Inam F, Reece MJ, Chlup Z, Dlouhy I, Shaffer MSP, et al. Carbon nanotubes: do they toughen brittle matrices? J Mater Sci. 2011;46(14):4770–9. doi:10.1007/s10853-011-5387-x.
Bokobza L, Zhang J. Raman spectroscopic characterization of multiwall carbon nanotubes and of composites. Express Polym Lett. 2012;6(7):601–8. doi:10.3144/expresspolymlett.2012.63.
Estili M, Kawasaki A, Pittini-Yamada Y, Utke I, Michler J. In situ characterization of tensile-bending load bearing ability of multi-walled carbon nanotubes in alumina-based nanocomposites. J Mater Chem. 2011;21(12):4272–8. doi:10.1039/c0jm03906c.
White AA, Best SM, Kinloch IA. Hydroxyapatite-carbon nanotube composites for biomedical applications: a review. Int J Appl Ceram Technol. 2007;4(1):1–13. doi:10.1111/j.1744-7402.2007.02113.x.
Das GK, Chan PPY, Teo A, Loo JSC, Anderson JM, Tan TTY. In vitro cytotoxicity evaluation of biomedical nanoparticles and their extracts. J Biomed Mater Res A. 2010;93A(1):337–46. doi:10.1002/jbm.a.32533.
Wallin R, Arscott E. A practical guide to ISO 10993-5: cytotoxicity. Med Device Diagn Ind Mag. 1998;20:96–8.
Pizzoferrato A, Ciapetti G, Stea S, Cenni E, Arciola CR, Granchi D, et al. Cell culture methods for testing biocompatibility. Clin Mater. 1994;15(3):173–90. doi:10.1016/0267-6605(94)90081-7.
Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small. 2008;4(1):26–49. doi:10.1002/smll.200700595.
Anselme K. Osteoblast adhesion on biomaterials. Biomaterials. 2000;21(7):667–81. doi:10.1016/S0142-9612(99)00242-2.
Detsch R, Guillon O, Wondraczek L, Boccaccini AR. Initial attatchment of rMSC and MG-63 Cells on patterned Bioglass® substrates. Adv Eng Mater. 2012;14(3):B38–44. doi:10.1002/adem.201180068.
Ghosh M, Chakraborty A, Bandyopadhyay M, Mukherjee A. Multi-walled carbon nanotubes (MWCNT): induction of DNA damage in plant and mammalian cells. J Hazard Mater. 2011;197:327–36. doi:10.1016/j.jhazmat.2011.09.090.
Misra SK, Ohashi F, Valappil SP, Knowles JC, Roy I, Silva SRP, et al. Characterization of carbon nanotube (MWCNT) containing P(3HB)/bioactive glass composites for tissue engineering applications. Acta Biomater. 2010;6(3):735–42. doi:10.1016/j.actbio.2009.09.023.
Chłopek J, Czajkowska B, Szaraniec B, Frackowiak E, Szostak K, Béguin F. In vitro studies of carbon nanotubes biocompatibility. Carbon. 2006;44(6):1106–11. doi:10.1016/j.carbon.2005.11.022.
Patlolla A, Knighten B, Tchounwou P. Multi-walled carbon nanotubes induce cytotoxicity, genotoxicity and apoptosis in normal human dermal fibroblast cells. Ethn Dis. 2010;20((1 Suppl 1)):S1-65–72.
Monteiro-Riviere NA, Nemanich RJ, Inman AO, Wang YY, Riviere JE. Multi-walled carbon nanotube interactions with human epidermal keratinocytes. Toxicol Lett. 2005;155(3):377–84. doi:10.1016/j.toxlet.2004.11.004.
Acknowledgments
The authors would like to thank European Union’s Seventh Framework Program managed by REA-Research Executive Agency http://ec.europa.eu/research/rea (Marie Curie Action, GlaCERCo GA 264526) and International Center for Young Scientists (ICYS) of National Institute for Materials Science (NIMS), Tsukuba, Japan for their support and funding for this research. Prof. M.J. Reece would like to acknowledge the support of Sunchon National University, South Korea, through the BK21 + programme.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Porwal, H., Estili, M., Grünewald, A. et al. 45S5 Bioglass®–MWCNT composite: processing and bioactivity. J Mater Sci: Mater Med 26, 199 (2015). https://doi.org/10.1007/s10856-015-5529-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-015-5529-9