
Abstract
This paper focused on the biodistribution of the cross-linked hyaluronic acid (HA-NPs) sub-micron particles in tumor-bearing mice. Solvent-non solvent method followed glutaraldehyde cross-linking utilized for the fabrication of HA-NPs. Size measurement and morphological analysis were performed by dynamic light scattering and electron microscopy, respectively and the size found to be in the range of 200–400 nm. In vitro viability in LNCaP cell line was assessed by water soluble tetrazolium assay after 24 h of exposure to sub-micron particles and no toxicity was found to higher concentration of 3 mg/mL. Internalization of particles in prostate cancer cell LNCaP were studied by confocal microscopy with FITC labeled submicron particles and involvement of hyaluronan receptor mediated uptake/endocytosis was confirmed by competitive assay. Biodistribution studies were performed in xenograft prostate cancer mice model with fluorophore labeled particles and monitored in tumoral parenchyma with strong fluorescence, meanwhile very less signal in liver, kidney and spleen while no fluorescence found in lung after 24 h of systemic administration; that shown ability of this HA based system to recognize cancer tissue. These result fetched that hyaluronic acid based system is selective for tumoral site and can be utilized to deliver bioactives in specific (targeting) and controlled (temporal) manner to cancerous tissue.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
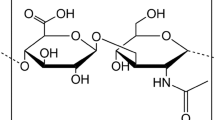
Hyaluronan (HA) is a linear, multifunctional glycosaminoglycan (GAG) composed of repeating disaccharide units of d-glucuronic acid and N-acetyl-d-glucosamine: [-b(1,4)-GlcUA-b(1,3)-GlcNAc-]n, constituent of the extracellular matrix (ECM) of connective tissue where it has a specific conformation useful to the maintenance of cellular micro environment and to regulate the binding of growth factors and cytokines to their receptors [1].
In cells, HA is produced by three types of enzymes called hyaluronan synthases (HAS1, HAS2, HAS3) at plasmatic membrane and degraded extracellularly by the action of hyaluronidases type 1/2, beta-glucuronidase and beta-N-acetylglucosaminidase [1].
HA participate in a wide variety of biological functions and seems to control angiogenic switch and activate intracellular pathways that are very important for cancer progression [2, 3] .
Major target of hyaluronan vector is CD44 a 85 kDa transmembrane receptor, involved in a variety of cellular functions including cell–cell aggregation, extravasation of leukocytes (leukocyte rolling), cell–matrix signalling, receptor-mediated internalization and cell migration.
In addition to these physiological properties, CD44 has been found to be overexpressed in most tumor cells and associated with tumor progression, as in the case of prostate tumors [4]. Preclinical studies have demonstrated a correlation among CD44-mediated endocytosis, hyaluronan degradation and tumor metastatic aggressiveness: the malignant cells that better internalize and degrade hyaluronan appear to be the most metastatic [2, 3] .
These experimental findings have been confirmed also in clinical studies conducted on different type of tumors. Studies on patients with non-Hodgkin’s lymphoma have demonstrated a direct relation between tumor progression and CD44v6 isoform expression suggesting this variant as an independent prognostic factor [5] .
Similar observations have been reported also for colorectal [6], gastric [7], pancreatic [8], renal [9], hepatocellular [10], cervical [11], ovarian [12], non-small lung [13], breast carcinoma [14] and melanoma [15], in which the presence of the isoforms, and in particular of CD44v6, was associated with advanced stage, unfavourable clinicopathological features and an adverse prognosis.
Among different types of micro- and nano-particles, currently under investigation, hyaluronan vectors have attracted considerable scientific and applicative interest [1, 16, 17].
The hydrophilic shells of these HA particles allow their prolonged circulation in the bloodstream, which may increase their probability of reaching tumor tissue after systemic administration in vivo [18, 19].
Furthermore, micro- and nano-particles have been reported to passively accumulate into tumor sites due to their abnormally leaky vasculature and lack of an effective lymphatic drainage system [20, 21]. As a consequence, anticancer drugs within the particles can be released inside tumor cells in controlled and intelligent manner [22–24]. Using this rationale, particles design for delivery of anticancer drugs may circulate for a long period of time in the bloodstream before reaching the tumor parenchyma by combination of three main factors: presence of peritumoral endothelial intercellular pores; decreased interstitial lymphatic drainage (Starling forces); absence of pericytes on tumor vascular endothelial cells, followed by cellular uptake at the target site due to receptor-mediated endocytosis [25].
The purpose of this paper was to demonstrate targeting efficiency of HA based system and its accumulation into the cancer cells after systemic administration, selectively. Sub micron particles prepared by non solvent emulsion method following chemical cross linking strategy. In vitro and in vivo assessment of chemically cross-linked sub-micron particles (HA-NPs) have carried out in prostate cancer cell line LNCaP and xenograft tumor bearing mouse model, respectively.
2 Materials and methods
2.1 Materials
Sodium hyaluronate (Mw: 200 kDa), purchased from Fidia (Italy), Formamide, Dimethyl sulfoxide (DMSO), Sodium carbonate, fluorescein isothiocyanate (FITC), Spectra/Por® Biotech Cellulose Ester Membrane (cut-off 300 kDa), Ethanol were obtained from Sigma-Aldrich Co. Water, used for synthesis and characterization, was purified by distillation, deionization, and reverse osmosis (Milli-Q Plus). All chemicals were analytical grade and used without further purification.
2.2 Preparation of HA-NPs cross-linked by glutaraldehyde
HA-NPs synthesis was performed by solvent–non solvent method [26, 27]. Briefly, a 0.5% w/vol HA sodium salt solution in bidistilled water was added dropwise (1 mL/min) to an equal volume of DMSO (1:1 ratio) under magnetic stirring (750 rpm).
HA-NPs were cross-linked adding glutaraldehyde in a reaction mixture (5 % vol./vol.). pH was adjusted to 4.5 with few drop of 0.1 M Hcl and reaction was allowed to proceed for 4 h under magnetic stirring (400 rpm). Then, cross-linked HA-NPs were purified by dialysis method using Spectra/Por® Biotech Cellulose Ester (CE) membrane with cut-off 300 kDa.
To further separate cross-linked and not cross-linked particles, we performed a purification step using a gel-filtration chromatography (GPC).
For recovery of cross-linked HA-NPs, we used a column Sigma Luer Lock, Non-Jacketed (ID × L (cm) 2.5 × 20) packed with Sepharose CL-2B resin (GE Healthcare Life Sciences), which allows a separation in range of size 7 × 104–4 × 107.
To facilitate the flow of eluent, Masterflex 7518-00 pump (Cole-Parmer Instrument Company) was connected to the head of the column while in the queue a pump ConstaMetric 3200 (Analytical PUMP LDC). As eluent was used distilled water (MilliQ). The eluted fractions were detected using a UV–Visible spectrometer setting the value of absorbance at 210 nm.
Since hydrodynamic radius of HA-NPs was influenced by solution ionic strength so we measured size distribution profile by DLS in both distilled water and 10 mM PBS solution. Cross-linked submicron particles were separated from uncross-linked HA by GPC and samples were freeze-dried, resuspended in 10 mM PBS, sonicated three times for 3 min each using a bath sonicator (Branson 5010) at 70 W and measured at DLS (1 mg/mL).
Hydrodynamic volumes of samples were measured using dynamic light scattering with a helium ion laser system Zetasizer Nanoseries ZEN3600 (Malvern Instruments, WR14 1XZ) which was operated at 633 nm. The scattered light was measured at an angle of 90° at temperature of 25 °C. Concentration of sub-micron particles was kept constant at 1 mg/mL. Each samples were measured three times.
Morphology of sub-micron particles was observed using environmental scanning electron microscopy (ESEM, FEI Company, Quanta 200) using an accelerating voltage of 10 keV, the chamber parameters were settled to 5 °C temperature and 3.60 Torr. The determination of cross-linked sub-micron particles mean diameter was performed on three different NPs preparations.
2.3 Labeling of cross-linked HA-NPs with FITC
Freeze-dried cross-linked HA-NPs were conjugated to FITC in organic solvent as described in literature [28], with little modification. We carried out the reaction with increasing time and temperature, without catalyst.
In brief, cross-linked HA-NPs (0.5 mg/mL) were dispersed in formamide under magnetic stirring (400 rpm) for 12 h. Reaction was performed at various times (24, 48, 72 and 96 h) adding a DMSO solution containing 8.5 mM FITC (final concentration). All the experiments were performed in triplicate.
Product was purified by precipitation in pure ethanol, filtered through a membrane filter (pore size: 0.45 μm, Millipore) and lyophilized.
HA-NPs conjugated to FITC (HA-F-NPs) were analyzed by infrared spectroscopy in attenuated total reflectance (IR–ATR mode) in order to verify the formation of the new thiocarbamate covalent bond between isothiocyanate FITC groups and carboxylic HA residues. HA-NPs degree of labeling (DOL) by FITC, defined as the percentage of FITC molecules bound to the carboxylic group of HA, was calculated spectrophotometrically at 481 nm.
2.4 Cell viability assay
Cell viability and proliferation were evaluated by a modified MTT method according to the manufacturer’s instructions (Dojindo Molecular Technologies Inc., Rockville, MD).
Briefly, quantification of mitochondrial dehydrogenase activity was achieved via the enzymatic conversion of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (Sigma-Aldrich) to a colored formazan product.
Since reduction in MTT occurs only in metabolically active cells, the level of activity refers to cell viability. 3T3 Swiss albino mouse fibroblast were seeded at density of 1 × 104 cells/well in a 24 multiwell in DMEM containing 10 % FBS, 1 % Pen–Strep, l-glutamine 2 mM and incubated at 37 °C in a 95 % air, 5 % CO2 in humidified atmosphere for 24 h.
Afterwards, growth medium was removed and replaced with fresh medium containing HA-NPs or HA-F-NPs at the following concentrations: 0.1, 0.3, 0.6, 1, 2 and 3 mg/mL, medium without sub-micron particles was used for control experiments. Each experimental point was performed in triplicate. Drugs cytotoxicity was calculated as medium value plus or minus standard deviation (SD) for each experimental point. MTT absorbance was calculated at 450 nm, (650 nm wavelength was used as turbidimetry control) using a Thermo Scientific spectrophotometer (Biomate 3).
2.5 In vitro cellular uptake of cross-linked HA-F-NPs
LNCaP cells were cultured in RPMI-1640 medium (Gibco) containing 15% FBS, 1% Pen–Strep, l-Glutamine 2 mM at 37 °C in a humidified 5 % CO2 atmosphere. For uptake experiments, 5 × 103 cells/well were seeded in 24 multiwell plate and allow to grown for 1 day in a serum-free culture medium (synchronization). After 1 day, the culture medium was replaced with one containing HA-F-NPs (0.3 mg/mL) and allowed to incubate for 4 h. Competitive assay was performed by incubation of cells with free HA (2 mg/mL).
Cells were then washed twice with PBS (pH 7.4) and fixed with cold methanol. Localization of HA-F-NPs were performed using a Confocal Microscope (C1 Nikon), in which the excitation and emission wavelengths were 492 and 518 nm, respectively.
2.6 In vivo biodistribution and tumor targeting characteristics of FITC HA-F-NPs
Tumor-bearing mice were prepared by injecting a suspension of LNCaP cells 1 × 106 in saline physiological solution (100 μL) into the subcutaneous dorsa of athymic nude mice (seven weeks old, 20–25 g). After fourteen days, HA-F-NPs were injected into the tail vein of the tumor-bearing mice at a dose of 5 mg/kg (corresponding to Human Equivalent Dose of 0,405 mg/kg) [29]. HA-F-NPs biodistribution was studied by fluorescence microscopy.
Xenograft mice were sacrificed at 4, 24 and 48 h after HA-F-NPs administration. Different organs and xenograft tumor implants were eradicated, transversely sectioned and slides (100 μm) of the middle region were obtained using a cryostat Leica CM1950.
Samples were mounted on glass coverslips and observed in fluorescence using 492 nm excitation and 518 nm emission wavelengths on a Nikon Eclipse Te 2000-s microscope. Images were collected with a 4× (Nikon plan fluor) lens.
Quantitative analysis of HA-F-NPs accumulation in tissues were determined using NIS-Elements BR2.30 software on selected region of interest (ROI). All values are expressed as mean ± SD for groups of three animals. To eliminate background fluorescence signal, we acquired fluorescence intensity signals from untreated organs and xenografts and subtract these values from the fluorescence intensity profile of treated mice.
3 Results
3.1 Synthesis and characterization of cross-linked HA-NPs
HA-NPs synthesised by solvent non solvent method followed by glutaraldehyde cross-linking, show a bimodal size distribution profile with two distinct peaks in the range 8–20 and 210–440 nm respectively (Fig. 1a). As observed in preliminary experiments, the peak in the range 8–20 nm was due to HA dissolved in water (data not showed) hence in Fig. 1a it indicates the presence of a not cross-linked HA fraction.

NPs size characterization. a DLS of cross-linked HA-NPs in PBS. b typical elution profile of cross-linked NPs on Sepharose CL-2B. c DLS of cross-linked and purified HA-NPs resuspended in PBS. d ESEM image of cross-linked HA-NPs after purification
The purification of the samples using the GPC method implies that larger particles are eluted earlier than the smaller ones (Fig. 1b), and integration of the relative peak obtained from chromatogram depicted approx 64 % of large HA nanoparticles (210–440 nm) eluted first and considered formation/cross-linking of sub-micron particles.
In Fig. 1c, the DLS analysis on GPC pooled fractions (4–35) was shown; the presence of a single peak in the range 210–440 nm demonstrates the separation between the two distinct populations of HA.
Moreover, presence of a single peak in Fig. 1c, after purification and sonication, indicates that the particle population (210–440 nm) was stable, vice versa a different size distribution profile was expected.
ESEM image in Fig. 1d indicate that the cross-linked HA-NPs have a spherical shape with an average diameter in the range 220 ± 50 nm.
The differences between light scattering and ESEM data were also due to the different hydration state of sub-micron particles.
3.2 Labeling of cross-linked HA-NPs with FITC
We have synthesized fluorescent HA-F-NPs by conjugating FITC to cross-linked HA-NPs with little modification. We got a DOL higher than reported in the literature with a maximum value of 0.6% (Fig. 2a).

Characterization of NPs-FITC, all experiments were performed in triplicate (n = 3). a Degree of labeling (DOL) in function of time. b FTIR–ATR mode spectra of FITC. c FTIR–ATR mode spectra of HA-NPs. d FTIR–ATR mode spectra of FITC-HA-NPs
The ATR–FTIR analysis confirmed the presence of the dye: in Fig. 2d we observe a peak at 2,000–2,270 cm−1 corresponding to isothiocyanate group (NCS) of FITC linked to HA, the stretching of the aromatic double bond, σ and π, at wavenumber 1,475 and 1,600 cm−1, the bending of aromatic C–H at wavenumber 690 cm−1 and the stretching of aromatic C–H at 3,030 cm−1.
We also observe characteristic signals of HA, such as a broad peak at 3,200–3,500 and 1,700–1,725 cm−1 (due to, stretching O–H and carboxyl groups absorptions, respectively) and a peak at 2,180 cm−1 due to the formation of thiocarbamate bound between FITC and carboxylic group of HA. All signals, clearly, showed the presence of FITC group on HA molecules (Fig. 2d).
3.3 Cytotoxicity studies of HA-NPs
The experiments of cell viability show that cross-linked HA-NPs are not toxic for cells. However, at high HA-NPs concentrations (2 and 3 mg/mL) a slight decrease of cell viability evident, probably, at high HA concentration, both the increase in solution viscosity and the polyelectrolyte nature of HA interfere with membrane transport mechanisms (Fig. 3). Moreover, the addition of FITC to our NPs system don’t affects significantly cell viability.

Cell vitality assay of cross-linked HA-NPs and cross-linked HA-F-NPs on 3T3 cell line
3.4 In vitro cellular uptake studies
Figure 4 shows results from HA-F-NPs uptake experiments. After two hours of contact, sub-micron particles are massively distributed in cytoplasm of cancer cells (Fig. 4a). In Fig. 4b the simultaneous administration of HA-F-NPs and a five times excess of free hyaluronic acid, give a marked inhibition of sub-micron particles cellular uptake, indicating an active internalization of HA-F-NPs due to HA receptors mediated endocytosis.

NPs cellular uptake. a fluorescence image of LNCaP cells after 2 h of contact with HA-F-NPs. b competitive assay, fluorescence image of LNCaP cells after 2 h of contact with HA-F-NPs and five times excess of free HA
Moreover, it seems that the presence of FITC on HA-NPs does not affect the binding of hyaluronate vector to the cellular target.
3.5 Biodistribution study in xenograft mouse model
In this experiment we study biodistribution of HA-F-NPs in xenograft mice through evaluation of fluorescence emission in various vital organs and tumor xenografts after intravenous administration.
As shown in Fig. 5, the fluorescence signal was absent in lung at any times, indicating that HA-F-NPs don’t accumulate in this organ, as confirmed also by semi-quantitative analysis (Fig. 6).

Fluorescence images of HA-F-NPs in xenograft mouse model after systemic administration

Time dependent fluorescence intensity profiles in different organs of cancer xenograft mouse model
In liver, fluorescence intensity increases with time up to 24 h (about 15 % of total fluorescence), evidently a slight reduction after 48 h of administration (Fig. 6).
Similar behaviour was observed in spleen and kidney cortex, fluorescence intensity was about 10 % at 24 h and also a slightly lesser at 48 h (Fig. 6).
Interestingly, the fluorescent signal for HA-F-NPs was stronger in the parenchyma of tumor site compared to other mice organs, resulting in about 60 % of total fluorescence at 24 h (Fig. 6).
4 Discussion
Due to its biocompatibility and biodegradability, HA has been extensively investigated as drug delivery system [16, 17]. In particular, most HA-drug conjugates have been developed for cancer chemotherapy as macromolecular prodrugs. These HA conjugates containing anticancer agents such as paclitaxel [30] and siRNA [31], exhibited enhanced targeting ability to the tumor and higher therapeutic efficacy compared to free anti-cancer agents.
The limitation of HA as a targeting carrier, rapid clearance from the circulation by the liver [32]. For HA to be a useful intravenous targeting carrier, strategies have been devised to reduce HA clearance from the blood. Degradation of HA by hyaluronidases can be reduced through chemical modifications such as attachment of HA to a noncarrier/drug [17] or crosslinking of individual HA polymer chains with the formation of nanogel particles [16].
The constraints applied to individual HA chains by their crosslinking or binding to the surface of more dense nanoparticles are believed to alter HA pharmacokinetic properties. However, it has been shown that HA nanoparticles can efficiently accumulate in tumors and be retained there for at least 2 days, whereas native HA is quickly cleared from the body within 1 h postinjection [33]. This behaviour might result from the increased circulation time and EPR effect as well as tumor selective binding of HA to CD44 receptor. In an other paper, Choi et al., using a mouse xenograft squamous carcinoma model, showed that self-assembled HA-NPs can accumulate into the tumor site by a combination of passive and active targeting mechanisms [18]. Moreover, the same authors, physically encapsulated irinotecan (an anticancer drug) into the hydrophobic cores of HA-NPs that exhibited an excellent antitumor activity in orthotopic colon cancer models, while showing a reduction in undesirable systemic toxicity [19]. Overall, the production of nanostructured materials from hyaluronic acid can tailor many properties of nano-scale drug delivery systems for specific applications: solubility (inherent hydrophilicity of HA), biodistribution (targeting to cancer cells), biocompatibility (electrical charge), biodegradability (binding/crosslinking density) and drug release (HA-drug spacer or physical interaction between a drug and a carrier) [16].
For these reason we prepared and studied chemical cross-linked hyaluronic acid sub-micron particles with an average diameter of 310 nm (Fig. 1d) which makes them responsive to EPR effect and permit a good accumulation in tumor tissue, as demonstrated by HA-F-NPs biodistribution studies in xenograft mice (Figs. 5, 6).
As known in the literature, tumors have pH values lower than normal tissue, hence, would seem misplaced the use of FITC (pH sensitive) for HA labeling. This choice has sprung by two aspects: reaction between FITC and HA carboxyl groups is simple, stable [34], well known and effective [28]; the fluorophore retains a large part of its fluorescence in the pH range 6.5–8 [34]. However, keeping in minds these observations, it is clear that the evaluation of fluorescence in vivo experiments is a semi-quantitative analysis.
In vitro study (Fig. 4) clearly showed the capacity of HA-F-NPs to accumulate in LNCaP prostate cancer cell line. Moreover, the uptake was mediated by HA membrane receptors as demonstrated by the absence of fluorescence signals inner cells in competitive experiment with free HA.
Biodistribution results indicate that HA-F-NPs accumulate with a high efficiency in xenograft tumor site (Figs. 5, 6). In addition, we observe that HA-F-NPs accumulate in time-dependent manner in liver, spleen and kidney cortex which may be due to cellular uptake of the HA-NPs by phagocytic cells of the reticuloendothelial system expressing Toll like receptors type 4 (TLR4) and by liver sinusoidal endothelial cells expressing the HARE receptor.
However, no fluorescence signal was detected in lung at any experimental times, showing that HA-F-NPs did not accumulate in pulmonary parenchyma.
Semi-quantitative analysis of fluorescence shows that, after 24 h, about 15 % of sub-micron particles were accumulated in the liver, 10 % in kidney and 10 % in the spleen.
The major signals were detected in xenograft implants with a specific fluorescence spots indicative of high HA-F-NPs accumulation. In particular, we observe that in each accumulation sites the maximum signals were detected after 24 h of systemic administration, while at 48 h the fluorescence signals decrease. Probably, in vital organs, this behaviour were due to physiological HA catabolism that is enhanced in tumor sites due to the hyper secretion of hyaluronidase [35].
Our results, together with literature data, suggest the use of cross-linked HA-NPs as a vector system for drug release at the aim to develop new anticancer therapies.
Further studies are in progress in order to bind anticancer drugs to sub-micron HA particles, mainly through pH sensitive chemical bonds.
References
Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information rich system. Eur J Cell Biol. 2006;85:699–715.
Zahalka MA, Okon E, Gosslar U, Holzmann B, Naor D. Lymph node (but not spleen) invasion by murine lymphoma is both CD44- and hyaluronate-dependent. J Immunol. 1995;154:5345–55.
Strobel T, Swanson L, Cannistra CA. In vivo inhibition of CD44 limits intra abdominal spread of a human ovarian cancer xenograft in nude mice: a novel role for CD44 in the process of peritoneal implantation. Cancer Res. 1997;57:1228–32.
Benitez A, Yates TJ, Lopez LE, Cerwinka WH, Bakkar A, Lokeshwar VB. Targeting hyaluronidase for cancer therapy: antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res. 2011;71:4085–95.
Stauder R, Eisterer W, Thaler J, Gunthert U. CD44 variant isoforms in non-Hodgkin’s lymphoma: a new independent prognostic factor. Blood. 1995;85:2885–99.
Mulder JW, Kruyt PM, Sewnath M, Oosting J, Seldenrijk CA, Weidema WF, Offerhaus GJ, Pals ST. Colorectal cancer prognosis and expression of exon-v6-containing CD44 proteins. Lancet. 1994;344:1470–2.
Mayer B, Jauch KW, Gunthert U, Figdor CG, Schildberg FW, Funke I, Johnson JP. De novo expression of CD44 and survival in gastric cancer. Lancet. 1993;342:1019–22.
Günthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13–24.
Terpe HJ, Storkel S, Zimmer U, Anquez V, Fischer C, Pantel K, Gunthert U. Expression of CD44 isoforms in renal cell tumors Positive correlation to tumor differentiation. Am J Pathol. 1996;148:453–63.
Endo K, Terada T. Protein expression of CD44 (standard and variant isoforms) in hepatocellular carcinoma: relationships with tumor grade clinicopathologic parameters p53 expression and patient survival. J Hepatol. 2000;32:78–84.
Kainz C, Kohlberger P, Sliutz G, Tempfer C, Heinzl H, Reinthaller A, Breitenecker G. Koelbl H splice variants of CD44 in human cervical cancer stage IB to IIB. Gynecol Oncol. 1995;57:383–7.
Uhl-Steidl M, Muller-Holzner E, Zeimet AG, Adolf GR, Daxenbichler G, Marth C, Dapunt O. Prognostic value of CD44 splice variant expression in ovarian cancer. Oncololgy. 1995;52:400–6.
Harada N, Mizoi T, Kinouchi M, Hoshi K, Ishii S, Shiiba K, Sasaki I, Matsuno S. Introduction of antisense CD44 cDNA down-regulates expression of overall CD44 isoforms and inhibits tumour growth and metastasis in highly metastatic colon carcinoma cells. Int J Cancer. 2001;91:67–75.
Kaufmann M, Heider KH, Sinn HP, von Minckwitz G, Ponta H, Herrlich P. CD44 variant exon epitopes in primary breast cancer and length of survival. Lancet. 1995;345:615–9.
Manten-Horst E, Danen EH, Smit L, Snoek M, Le Poole IC, Van Muijen GN, Pals ST, Ruiter DJ. Expression of CD44 splice variants in human cutaneous melanoma and melanoma cell lines is related to tumor progression and metastatic potential. Int J Cancer. 1995;64:182–8.
Ossipov DA. Nanostructured hyaluronic acid-based materials for active delivery to cancer. Expert Opin Drug Deliv. 2010;7:681–703.
Platt VM, Szoka FC Jr. Anticancer therapeutics: targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol Pharm. 2008;5:474–86.
Choi KY, Chung H, Hyun K, Hong M, Yoon Y, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106–14.
Choi KY, Jeon EJ, Yoon HY, Lee BS, Na JH, Min KH, Kim SY, Myung SJ, Lee S, Chen X, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials. 2012;33:6186–93.
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92.
Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62:90–9.
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60.
Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006;58:1532–55.
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–26.
Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrinconjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–83.
Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25:2843–9.
Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2:8–21.
de Belder AN, Wik KO. Preparation and properties of fluorescein-labeled hyaluronate. Carbohydr Res. 1975;44:251–7.
Shannon RS, Minakshi N, Nihal A. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61.
Auzenne E, Ghosh SC, Khodadadian M, Rivera B, Farquhar D, Price RE, et al. Hyaluronic acid-paclitaxel: antitumor efficacy against CD44(þ) human ovarian carcinoma xenografts. Neoplasia. 2007;9:479–86.
Lee H, Mok H, Lee S, Oh YK, Park TG. Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels. J Control Release. 2007;119(2):245–52.
Harris EN, Kyosseva SV, Weigel JA, Weigel PH. Expression, processing, and glycosaminoglycan binding activity of the recombinant human 315-kDa hyaluronic acid receptor for endocytosis (HARE). J Biol Chem. 2007;282(5):2785–97.
Choi KY, Min KH, Na JH, et al. Self-assembled hyaluronic acid nanoparticles as a potential drug carrier for cancer therapy: synthesis, characterization, and in vivo biodistribution. J Mater Chem. 2009;19(24):4102–7.
Kurtzhals P, Larsen C, Johansen M. High performance size-exclusion chromatographic procedure for the determination of fluoresceinyl isothiocyanate dextrans of various molecular masses in biological media. J Chromatogr. 1989;491:117–27.
Dacai L, Pearlment E, Diaconu E, Kun G, Hiroshi M, HaqqI S, Sanford M, Willson J, Sun SM. Expression of hyaluronidase by tumor cells induces angiogenesis in vivo. Proc Natl Acad Sci. 1996;93:7832–7.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rosso, F., Quagliariello, V., Tortora, C. et al. Cross-linked hyaluronic acid sub-micron particles: in vitro and in vivo biodistribution study in cancer xenograft model. J Mater Sci: Mater Med 24, 1473–1481 (2013). https://doi.org/10.1007/s10856-013-4895-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-013-4895-4