
Abstract
A new strategy for assembling multifunctional nanocomposites with magnetic particles and amino dendrimers was reported. In this strategy, the amino terminated PAMAM G5.0 and Fe3O4 NPs prepared by co-deposition method and further modified by aminosilane by two sol–gel processes were combined with the hydrophilic spacer of PEG dicarboxylate by amidation. The nanocomposites were characterized by means of X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), atom force microscopy (AFM), superconducting quantum interference device (SQUID) magnetometer, and hydrophilicity analysis. The results showed that the multifunctional nanocomposites were spherical with the mean diameter of 180 nm and exhibited good dispersion and hydrophilicity. The new strategy put forward here provides an effective route to functionalizing Fe3O4 NPs with various amino dendrimers for drug and gene delivery as well as biological detection.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Magnetic nanoparticles have attracted intense attention in biomedical applications [1], such as separation of biomacromolecules [2], magnetic resonance imaging [3], biological labels [4], and targeted drug delivery [5–7]. Dendrimers as a new class of macromolecules are known for their three-dimensional, monodispersed, highly branched, nanoscopic architecture with a large number of reactive end groups [8, 9]. Recently, dendrimers have been used successfully in the field of biomedicine [10], in particular for their use as drug and gene delivery [11], cancer diagonosis [12], and sensors [13]. Assembly of inorganic nanoparticles with dendrimers has received increasing attention due to the combination of the properties of inorganic nanoparticles and dendrimers [14–17]. Multifunctional nanocomposites combining with magnetism of magnetic particles and physicochemical properties of dendrimers have become an ideal candidate for applications in the field of biomedicine. For the polyamidoamine (PAMAM) dendrimers have different terminal functionality (termination at a half generation gives carboxyl terminal groups; whereas full generation is terminated by amine functions), in accordance, the nanocomposites can also be divided into carboxyl- and amino- terminated types. Strable et al. [18] and Shi et al. [19] synthesized carboxyl-terminated polyamidoamine (PAMAM) dendrimer-iron oxide nanocomposites by a template technique for magnetic resonance imaging applications. The multifunctional nanocomposites found to have unusually high T1 and T2 NMR relaxivities are currently being explored as the basis for a new generation of contrast agents for magnetic resonance imaging applications [20]. Wang et al. [21] reported the synthesis of the amino-terminated nanocomposites by electrostatic layer-by-layer self-assembly technique for specific targeting of cancer cells. The amino-terminated multifunctional nanocomposites with large numbers of functional amino groups outside can further react with some ligands or adsorb DNA plasmid. Therefore, the amino-terminated nanocomposites may also have unique potential in drug and gene delivery.
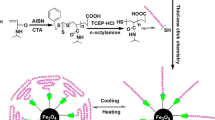
In this study, we develop a new strategy to assemble amine PAMAM dendrimers with magnetic nanoparticles. For PAMAM dendrimers need to improve water miscibility for in vivo applications [22], a hydrophilic spacer was introduced in the process of assembly so as to increase the hydrophilicity. Because polyethylene glycol (PEG) has been shown to be biocompatible, non-immunogenic, non-antigenic, protein resistant and with high solubility in cell membranes [23, 24], PEG was selected as the skeleton of the hydrophilic spacer for assembling dendrimers and iron oxide particles. As summarized in Fig. 1, first iron oxide particles stabilized by poly(N-vinyl-2-pyrrolidone) (PVP) were synthesized by a co-deposition method. Then the magnetic particles were further modified by an aminosilane using two sol–gel processes. Finally, the magnetic iron oxide nanoparticles functionalizing by amino groups and the PAMAM G5.0 dendrimers terminated by amino groups were combined with the hydrophilic spacer of PEG biscarboxylate by amidation to form nanocomposites. Such nanocomposites showed excellent dispersion and hydrophilicity, which are the critical factors for in vivo applications. This strategy has the following advantages: (1) the method presented here may be extended to combine systems of other metal oxide and different types of dendrimers include polylysine dendrimers and polypropylene imine dendrimers, (2) the additional advantage for the strategy is the silane coating for the surface modification of magnetic particles. Because silane-coated nanoparticles have been extensively studied over the past decade, some fluorescent dye or quantum dots and pharmaceutical molecules can be easily incorporated into the silane shell [25–28]. So the new strategy to assemble the amine PAMAM dendrimers with magnetic nanoparticles would be beneficial to widen the applications of magnetic nanoparticles and dendrimers in drug and gene delivery as well as biological detection.

Schematic illustration of the synthesis procedure for nanocomposite
2 Experimental
2.1 Materials
PEG biscarboxylate, PAMAM dendrimers (G5.0), Tetraethylorthosilicate (TEOS, >99%) and 3-aminopropyl-triethoxy silane (APS, >95%) were ordered from Sigma-Aldrich. Ethylenediamine (≥99%), methyl acrylate (≥99.8%), methanol (≥99.8%), absolute ethanol (≥99.7%), ammonia (30%), toluene (≥98%), and acetone (≥99.5%) were all purchased from SCRC. Ltd. N,N-dimethyl-formamide (DMF, ≥99%), O-Benzotriazole-N,N,N′,N′- tetramethyl-uronium-hexafluorophosphate (HBTU, ≥99%), 1-Hydroxybenzotrizole (HOBT, ≥99%) were purchased from GL Biochem Ltd (Shanghai). Water used in the experiment was distilled. Toluene was redistilled and kept anhydrous. Other chemicals were used directly without further purification.
2.2 Synthesis of PVP-stabilized iron oxide nanoparticles (MP)
Briefly, 5.8 g of PVP was added to 20 ml of distilled water under magnetic stirring. After the total dissolution of the PVP, 5 ml of 3 M sodium hydroxide solution was added to the solution. Then 2.5 ml of 0.5 M ammonium ferrous sulfate and 5 ml of 0.5 M iron sulfate solution were mixed and dropped to the solution above under magnetic stirring and a nitrogen protection. The system was maintained under magnetic stirring at 70°C for 1 h and subsequently the heating was turned off. A very stable colloidal dispersion was formed with this procedure. The PVP-stabilized iron oxide nanoparticles were separated by centrifugation at 9000 rpm for 10 min.
2.3 Synthesis of magnetic nanoparticles functioned by amino silane (MPA)
Then the sol–gel processes were carried out via the modified Stöber method [29]. Under continuous mechanical stirring 37 ml of PVP-iron oxide solution (3 mg/ml) and 150 ml absolute ethanol were mixed together. Then 0.5 ml of ammonia solution and 0.625 ml of tetraethylorthosilicate (TEOS) were added to the solution above. The reaction was allowed to proceed at 35°C in water bath for 12 h under continuous stirring. The second sol–gel process was performed as following. 0.1 g of magnetic particles was added to 100 ml of toluene solution. Then 0.65 ml of 3-aminopropyl-triethoxy silane (APS) was added to solution above under vigorous stirring. This hydrolysis reaction of APS proceeded for 24 h at 25°C to form MPA particles. Finally, the particles were isolated by centrifugation at 9000 rpm for 10 min.
2.4 Synthesis of magnetic nanoparticles combined with the spacer PEG dicarboxylic acid (MPA-PEG) and nanocomposites (MPA-PEG-PAMAM)
First, the magnetic nanoparticles and PEG dicarboxylic acid were coupled together. Briefly, 0.1 g of MPA and 1.87 g of PEG dicarboxylic acid were mixed together in 10 ml of N,N-dimethyl-formamide (DMF) containing 215 μl of triethylamine. Under mechanical stirring, 20 ml of DMF dissolving 0.593 g of HBTU and 0.241 g of HOBT was dropped to the mixture. The amidation reaction proceeded at 25°C for 48 h under tempestuously stirring to give MPA-PEG. With similar amidation reaction, 0.3 g of PAMAM G5.0 dendrimers (amine terminated) and 0.1 g of MPA-PEG were coupled to give MPA-PEG-PAMAM nanocomposites.
3 Methods for characterization
Powder XRD measurements were performed at room temperature with a D/max 2400 X-diffraction meter equipped with a Cu Kα radiation source (λ = 0.154056 nm). The patterns were obtained within the 2θ range of 20–80° in step mode with collection time of 6 min.
TEM images were obtained using a JEOL 2010 microscope operating at 200 kV. Samples were prepared by depositing a drop of the diluted nanocomposites suspension on 300 mesh carbon support films and drying the grids under vacuum for 2 h.
XPS experiments were performed using a commercial system (Thermo VG ESCALAB250 with Al Kα X-ray source, 1486.6 eV photons) with a base pressure of 1 × 10−7 mbar. The powder samples were mounted onto standard VG sample studs with double-sided adhesive tapes prior to each experiment. In order to observe the atoms on the surface layer in their different chemical environments, the individual peaks in the XPS spectrum were de-convoluted according to the method reported by Wei et al. [30]. The binding energy (BE) of the core level C 1 s peak was set at 285.0 eV to compensate for surface-charging effects of PVP [31]. The Shirley background was subtracted for all element peaks before curve fitting. The experimental spectra were fitted into components of Gaussian line shape. The surface elemental compositions were determined by the ratios of peak areas corrected with empirical sensitivity factors.
Quantum Design MPMS-XL superconducting quantum interference device (SQUID) magnetometer was used for function characterization. Zero-field-cooled (ZFC) magnetizations were measured by cooling the sample from room temperature down to about 30 K and then by increasing the temperature from 30 K up to 315 K in zero field. Then field -cooled (FC) magnetizations were measured by cooling the sample from 315 K to 30 K in an applied field of 100 Oe. The magnetization data were recorded in this cycle. To measure the hysteresis loops, a field of 20 kOe were applied to the sample and gradually decreased to 0 followed by field reversal and gradually increased to −20 kOe. The sample was prepared by filling a gelatin capsule with magnetic powders, which was subsequently sealed with Paraffin.
Samples for AFM analysis were prepared by dropping the products ethanol solutions (0.1 mg/ml) onto the center of a freshly split untreated silica disk. Excess solution was removed by absorption onto filter paper and the silica surface was further dried at room temperature before imaging. The image mode was set to tapping mode and the average scanning speed was 5 Hz.
The hydrophilicity of nanocomposites was evaluated by measuring the dispersion rate (Fs) and time (T) relation developed by Ren et al. [32]. The nanocomposites dispersed in methanol were used as a control. The samples were prepared by mixing 0.125 mg magnetodendrimer with 50 ml water or 50 ml methanol to obtain a final nanocomposite concentration of 2.5 mg/l. After ultrasonic for 30 s, the nanocomposites solutions of 50 ml were divided into five. For each sample, we removed half of the pellucid supernatant fluid at 2 h interval. Ten hours later, all the samples and the supernatant fluid were dried in the vacuum oven for 72 h. By weighing the nanocomposites in each sample (W′) and supernatant fluid (Ws), we obtained the weight of dispersed nanocomposites (2Ws) and the weight of all nanocomposites (W′ + Ws). The dispersion rate (Fs) was calculated as a percent ratio of 2Ws and (W′ + Ws). The values of Fs for nanocomposites at 0 h were regarded as 100%. By this method, series values of Fs for 2, 4, 6, 8, and 10 h were obtained.
4 Results and discussion
4.1 Synthesis and characterization of nanocomposites
The XRD pattern of the iron oxide nanoparticles stabilized by PVP shown in Fig. 2a matches that for Fe3O4 listed in XRD standard card (No: 85-1436). This result confirms the formation of the crystalline structure of the iron oxide nanoparticles. The lattice parameter calculated, according to the (440) diffraction peak, of the as-prepared cubic (Fd3m) Fe3O4 nanoparticles is 8.344 Å, which is smaller than 8.393 Å of the Fe3O4 raw powders. This difference suggests that crystal defects exist in the Fe3O4 lattice. The average size of the crystalline Fe3O4 particles is about 6.5 nm, as calculated by Scherrer equation. Figure 2b showed TEM images of the iron oxide nanoparticles stabilized by PVP, from which the formation of the core-shell structure and the dispersion of the magnetic nanoparticles were clearly observed.

XRD spectrum and TEM image of iron oxide particles stabilized by PVP
Once the synthesis of the magnetic nanoparticles had been confirmed by XRD spectroscopy and TEM, the PEG dicarboxylic acid and PAMAM dendrimers were immobilized on the nanoparticles following the scheme outlined in Fig. 1. As XPS can provide information on the chemical states at the surface of the material exposed to the incident X-rays and the part of the material where the changes happened during the procedures of the sol–gel process and the amidation reaction, XPS was selected to confirm the synthesis of the nanocomposite [33]. The survey spectra of all the MPA, MPA-PEG, and MPA-PEG-PAMAM are similar in general outline as shown in Fig. 3a. However, the changes of peak’s areas provide useful information about the surface loading of the PEG and the PAMAM onto MPA. On attaching the PEG and PAMAM to the MPA surface, a greater degree of carbon (and nitrogen) relative to MPA were observed. Data in Table 1 show that the C/Si ratio increases from 1.75 for MPA to 2.76 for MPA-PEG and 3.05 for MPA-PEG-PAMAM, respectively. It was also of interest to determine N/C ratios (Table 1). For the amino propyl groups and dendritic branching with higher N/C ratio whereas PEG spacer with lower C/N ratio, therefore, the N/C ratio varies from 0.14 for MPA to 0.083 for MPA-PEG and to 0.14 for MPA-PEG-PAMAM. This proved that the PEG spacer and PAMAM dendrimers had been attached to the surface of MPA. Figure 3b shows the deconvoluted N 1s spectra for the MPA, MPA-PEG, and MPA-PEG-PAMAM. By identification of the functional groups present on the surface, the N 1 s spectrum of the MPA could be decomposed into two components at 399.5 and 401.6 eV, which correspond to NH2 and NH3 + [33]. Following loading of the PEG spacer, states characteristic of RCNH (399.0 eV), and OCONH (400.3 eV) are visible in the N 1 s spectrum. Similar deconvolution of the MPA-PEG-PAMAM reveals N 1 s states consistent with NH2 at 399.7 eV.

XPS spectra of MPA, MPA-PEG, and MPA-PEG-PAMAM a the survey spectra, b the deconvoluted N 1s spectra
4.2 Dispersibility analysis
The dispersibility of nanocomposites is critical for the application of nanocomposites. Therefore, AFM technique was undertaken to determinate the dispersion of nanocomposites before and after the sol–gel processes and amidations. The AFM image in Fig. 4a shows that the iron oxide nanoparticles with the mean diameter of 26 nm are well dispersed. This may be attributed to the stabilization of PVP when the iron oxide formed. It is the stabilization that prevents the agglomeration of the nanoparticles. After the two sol–gel processes were performed, the synthesized MP with the mean diameter of 70 nm and MPA particles with the mean diameter of 85 nm were well dispersed too (as shown in Fig. 4b, c). After the amidations were performed, the MPA-PEG with the mean diameter of 118 nm and MPA-PEG-PAMAM particles with the mean diameter of 180 nm still exhibited good dispersion as shown in Fig. 4d, e. However, there is great difference between the images of MPA-PEG particles and MPA-PEG-PAMAM ones: (1) the difference in diameter can be due to the fact that MPA-PEG particles may cross link other MPA-PEG particles in the amidation with PAMAM dendrimers and form much larger MPA-PEG-PAMAM particles in diameter, (2) the difference in surface state can be due to that the highly branched structure of PAMAM dendrimers leads MPA-PEG-PAMAM to rise and fall on the surface as shown in Fig. 4f. It is interesting that the crosslinking during the amidations can be controlled by the amount of linker reagents. For example, the excessiveness of PEG dicarboxylic acid in the amidation greatly reduced the crosslinking between MPA particles as shown in Fig. 4d. On the contrary, insufficiency of PAMAM dendrimers in the amidation resulted of the crosslinking between MPA-PEG particles and the forming of large numbers of MPA-PEG-PAMAM-PEG-MPA particles. When the crosslinking stopped, some steric exclusion groups-PAMAM dendrimers were left on the outside surface of MPA-PEG-PAMAM particles. For loading high steric exclusion groups, the agglomeration of the magnetic particles was effectively prevented so that the final nanocomposites were well dispersed as shown in Fig. 4e.

AFM images of a MP (2 × 2 μm2), b MP particles coated by SiO2, 2 × 2 μm2, c MPA (2 × 2 μm2), d MPA-PEG (2 × 2 μm2), e MPA-PEG-PAMAM (2 × 2 μm2), f 3D top-view image of MPA-PEG-PAMAM (2 × 2 μm2)
4.3 Hydrophilicity analysis
Hydrophilicity of nanocomposites is one of the important properties for in vivo applications. It is well known that the PAMAM dendrimers are high methanol miscible but low water miscible, and they have a tendency to form aggregates deposition in water. So we adopt hydrophilic PEG spacer to increase the hydrophilicity of nanocomposites. In order to verify the role of PEG spacer, we compared the dispersion behavior of nanocomposites in methanol and in water. From the dispersion rate (Fs) and time (T) measurement of nanocomposites in water and in methanol shown in Fig. 5, we could observe that magnetodendrimers exhibited better dispersion behavior and the tendency of deposition was lowered in water. From this, we suggest that the nanocomposites are hydrophilic, which results from the contribution of hydrophilic PEG spacer.

The hydrophilicity measurement of nanocomposites
4.4 Magnetic properties
The zero-field-cooled (ZFC) and field-cooled (FC) temperature dependence of the magnetization for the MP sample was measured at a magnetic field of 100 Oe in a temperature range between 30 and 315 K. From Fig. 6a, it is clear that the ZFC and FC curves for the MP coincided at high temperatures, but show a bifurcation as decreasing the temperature. According to the calculation from XRD data by Scherrer equation and the TEM observation, the average size of the Fe3O4 particles was about 6.5 nm, which is smaller than their super paramagnetic critical size [34]. The maximum in the ZFC curve indicated that the blocking temperature (TB) for MP was 135 K. According to the super paramagnetic theory, the particle will have the super paramagnetic character above 135 K. It should be noticed that the magnetic properties of nanocomposites are contributed by MP. The hysteresis loops for MP and nanocomposites measured at 295 K were shown in Fig. 6b. The value of coercivity for MP and nanocomposites were 11.2 and 30.4 Oe, respectively, which can be caused by the aggregated particles. At room temperature, the Fe3O4 particles with a single magnetic domain may exhibit super paramagnetism, whereas the aggregated particles exhibit ferromagnetism. The saturation magnetization (Ms) decreased from 21.6 emu/g to 8.4 emu/g with increasing the non-magnetic organic components, which can reduce the total magnetization to a different extent [35]. Such a moderate magnetization can ensure an effective magnetic target for the delivery of drugs and gene.

a Zero-field-cooled (ZFC) and field-cooled (FC) temperature dependence of the magnetization for MP. b Hysteresis loops for MP and nanocomposites at 295 K
5 Conclusions
We have reported an effective strategy for assembling nanocomposites with Fe3O4 NPs and various amino dendrimers. The hydrophilic spacer-PEG dicarboxylate was selected to combine the iron oxide particles functioned by aminosilane and the amino PAMAM dendrimers. The nanocomposites with a mean diameter of 180 nm were well dispersed and hydrophilic. The amino terminal groups of the nanocomposites provide the nanocomposites a high potential application for drug and gene delivery.
References
Zhang ZD. Nanocapsules. In: Nalwa HS, editor. Encyclopedia of nanoscience and nanotechnology, vol 6. Stevenson Ranch, California: American Scientific Publishers; 2004. p. 77–160.
HW Gu, KM Xu, CJ Xu, B Xu, Biofunctional magnetic nanoparticles for protein separation and pathogen detection. Chem Commun. 2006; 941–9. doi:10.1039/b514130c.
Oscar BM, María PM, Pedro T, Jesus RC, Pierre B, Martín S, et al. Fe-based nanoparticles metallic alloys as contrast agents for magnetic resonance imaging. Biomaterials. 2005;26:5695–703. doi:10.1016/j.biomaterials.2005.02.020.
Xie HY, Zuo C, Liu Y, Zhang ZL, Pang DW, Li XL, et al. Cell-targeting multifunctional nanospheres with both fluorescence and magnetism. Small. 2005;1:506–9. doi:10.1002/smll.200400136.
Yang Y, Jiang JS, Du B, Gan ZF, Qian M, Zhang P. Preparation and properties of a novel drug delivery system with both magnetic and biomolecular targeting. J Mater Sci: Mater Med. 2009;20:301–7. doi:10.1007/s10856-008-3577-0.
Gou ML, Qian ZY, Wang H, Tang YB, Huang MJ, Kan B, et al. Preparation and characterization of magnetic poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) microspheres. J Mater Sci: Mater Med. 2008;19:1033–41. doi:10.1007/s10856-007-3230-3.
Tanaka H, Sugita T, Yasunaga YJ, Shimose SJ, Deie M, Kubo T, et al. Efficiency of magnetic liposomal transforming growth factor-β 1 in the repair of articular cartilage defects in a rabbit model. J Biomed Mater Res. 2005;73A:255–63. doi:10.1002/jbm.a.30187.
Aulenta F, Hayes W, Rannard S. Dendrimers: a new class of nanoscopic containers and delivery devices. Eur Polym J. 2003;39:1741–71. doi:10.1016/S0014-3057(03)00100-9.
Boas U, Heegaard PMH. Dendrimers in drug research. Chem Soc Rev. 2004;33:43–63. doi:10.1039/b309043b.
Paleos CM, Tsiourvas D, Sideratou Z. Molecular engineering of dendritic polymers and their application as drug and gene delivery systems. Mol Pharmacol. 2007;4:169–88. doi:10.1021/mp060076n.
Kim TI, Seo HJ, Choi JS, Jang HS, Baek JU, Kim K, et al. PAMAM-PEG-PAMAM: novel triblock copolymer as a biocompatible and efficient gene delivery carrier. Biomacromolecules. 2004;5:2487–92. doi:10.1021/bm049563j.
Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR Jr. PAMAM dendrimer-based multifunctional conjugate for cancer therapy: synthesis, characterization, and functionality. Biomacromolecules. 2006;7:572–9. doi:10.1021/bm0506142.
Myc A, Majoros IJ, Thomas TP, Baker JR Jr. Dendrimer-based targeted delivery of an apoptotic sensor in cancer cells. Biomacromolecules. 2007;8:13–8. doi:10.1021/bm060815l.
Shi XY, Wang SH, Sasha M, Mary EVA, Bi XD, Lee IH, et al. Dendrimer-entrapped gold nanoparticles as a platform for cancer-cell targeting and imaging. Small. 2007;3:1245–52. doi:10.1002/smll.200700054.
Scott RWJ, Datye AK, Crooks RM. Bimetallic palladium-platinum dendrimer-encapsulated catalysts. J Am Chem Soc. 2003;125:3708–9. doi:10.1021/ja034176n.
Jiang YJ, Jiang JH, Gao QM, Ruan ML, Yu HM, Qi LJ. A novel nanoscale catalyst system composed of nanosized Pd catalysts immobilized on Fe3O4@SiO2–PAMAM. Nanotechnology. 2008;19:1–6. doi:10.1088/0957-4484/19/7/075714.
Mandal D, Maran A, Yaszemski MJ, Bolander ME, Sarkar G. Cellular uptake of gold nanoparticles directly cross-linked with carrier peptides by osteosarcoma cells. J Mater Sci: Mater Med. 2009;20:347–50. doi:10.1007/s10856-008-3588-x.
Strable E, Bulte JWM, Moskowitz B, Vivekanandan K, Allen M, Douglas T. Synthesis, characterization, and intracellular uptake of carboxyl-terminated poly(amidoamine) dendrimer-stabilized iron oxide nanoparticles. Chem Mater. 2001;13:2201–9. doi:10.1021/cm010125i.
Shi XY, Thomas TP, Myc LA, Kotlyar A, Baker JR Jr. Synthesis, characterization, and intracellular uptake of carboxyl-terminated poly(amidoamine) dendrimer-stabilized iron oxide nanoparticles. Phys Chem Chem Phys. 2007;9:5712–20. doi:10.1039/b709147h.
Bulte JWM, Douglas T, Witwer B, Zhang SCh, Strable E, Lewis BK, et al. Magnetodendrimers allow endowsomal magnetic labeling and in vivo stracking of stem cells. Nat Biotechnol. 2001;19:1141–7. doi:10.1038/nbt1201-1141.
Wang SH, Shi XY, Antwerp MV, Cao ZY, Swanson SD, Bi XD, et al. Dendrimer-functionalized iron oxide nanoparticles for specific targeting and imaging of cancer cells. Adv Funct Mater. 2007;17:3043–50. doi:10.1002/adfm.200601139.
Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB, Demanuele A. The influence of surface modification on the cytotoxicity of PAMAM dendrimers003. Int J Pharm. 2003;252:263–6. doi:10.1016/S0378-5173(02)00623-3.
Yamazaki M, Ito T. Deformation and instability of membrane structure of phospholipid vesicles caused by osmophobic association: mechanical stress model for the mechanism of poly(ethylene glycol)-induced membrane fusion. Biochemistry. 1990;29:1309–14. doi:10.1021/bi00457a029.
Boni LT, Hah JS, Hui SW, Mukherjee P, Ho JT, Jung CY. Aggregation and fusion of unilamellar vesicles by poly(ethylene glycol). Biochem Biophys Acta-Biomembranes. 1984;775:409–18. doi:10.1016/0005-2736(84)90387-0.
Yoon TJ, Kim JS, Kim BG, Yu KN, Cho MH, Lee JK. Multifunctional nanoparticles possessing a “magnetic motor effect” for drug or gene delivery. Angew Chem. 2005;44:1092–5. doi:10.1002/anie.200461910.
Guo J, Yang WL, Deng YH, Wang CC, Fu SK. Organic-dye-coupled magnetic nanoparticles encaged inside thermoresponsive PNIPAM microcapsules. Small. 2005;1:737–43. doi:10.1002/smll.200400145.
Zhang Y, Wang SN, Ma S, Guan JJ, Li D, Zhang XD, et al. Self-assembly multifunctional nanocomposites with Fe3O4 magnetic core and CdSe/ZnS quantum dots shell. J Biomed Mater Res. 2008;85A:840–6. doi:10.1002/jbm.a.31609.
Souza KC, Ardisson JD, Sousa EMB. Study of mesoporous silica/magnetite systems in drug controlled release. J Mater Sci: Mater Med. 2009;20:507–9. doi:10.1007/s10856-008-3592-1.
Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica sphere in micro size range. J Colloid Interface Sci. 1968;26:62–9. doi:10.1016/0021-9797(68)90272-5.
Wei XL, Fahlman M, Epstein KJ. XPS study of highly sulfonated polyaniline. Macromolecules. 1999;32:3114–7. doi:10.1021/ma981386p.
Jiang P, Zhou JJ, Li R, Gao Y, Sun TL, Zhao XW, et al. PVP-capped twinned gold plates from nanometer to micrometer. J Nanopart Res. 2006;8:927–34. doi:10.1007/s11051-005-9046-5.
J. Ren, J. Shen, S.C. Lu. In: Xing T, editor. Dispersion science and technology of particles. Beijing: Chemical Industry Press; 2005. p. 175.
Driffield M, Goodall DM, Klute AS, Smith DK, Wilson K. Synthesis and characterization of silica-supported L-lysine-based dendritic branches. Langmuir. 2002;18:8660–5. doi:10.1021/la0203842.
Lee J, Isobe T, Senna M. Preparation of ultrafine Fe3O4 particles by precipitation in the presence of PVA at high pH. J Colloid Interface Sci. 1996;177:490–4. doi:10.1006/jcis.1996.0062.
Ding Y, Hu Y, Zhang LY, Chen Y, Jiang XQ. Synthesis and magnetic properties of biocompatible hybrid hollow spheres. Biomacromolecules. 2006;7:1766–72. doi:10.1021/bm060085h.
Acknowledgments
This work was supported by National Natural Science Foundation of China under Grant No. 50331030 and 50831006.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, Y., Liu, JY., Yang, F. et al. A new strategy for assembling multifunctional nanocomposites with iron oxide and amino-terminated PAMAM dendrimers. J Mater Sci: Mater Med 20, 2433–2440 (2009). https://doi.org/10.1007/s10856-009-3808-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-009-3808-z