
Abstract
Hydrogels are increasingly used in medicine due to their potential to be delivered into the body in a minimally invasive manner and to be gelated at the site of introduction subsequently. The aim of this study was to develop a novel injectable and in situ-forming gel composite (GC) comprised of calcium alginate hydrogel and nano-hydroxyapatite/collagen (nHAC), assess its rheological, mechanical and in vitro degradable properties, and discuss the gelation mechanism. Injectable property test showed that the injectability of GC was tunable. Rheological results indicated that three phases of pre-gel, sol–gel phase transformation and post-gel could be found in the process of gelation. The compressive elastic modulus (E) and shear modulus (G) are in the range of 17.0–56.0 kPa and 24.7–55.0 kPa, respectively. During the in vitro degradation, the wet weight increased in the first week, then declined in the following 3 weeks, but the dry weight lost continuously during whole study. Meanwhile, the surface changed greatly after 2 weeks, but samples did not break down up to 28 days. These data indicate that GC exhibits controllable initial setting time and final setting time, tunable injectability, which provides a possible injectable material for bone repair and bone tissue engineering.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
With the increasing popularity of minimally invasive techniques, injectable materials became one of the major hotspots in biomaterial field. The injectable scaffolds could shorten the surgical operation time, bring compliance and comfort to patients for non- or minimal invasive surgery. in situ forming scaffolds allows patients to recover rapidly in a cost-effective manner [1, 2].
Hydrogels, as injectable scaffolds, are currently used or have potential applications in tissue engineering as well as drug delivery systems [3–6]. Hydrogel scaffolds are usually made of polymers (e.g., PPF [7–10]) or biomacromolecules (e.g., chitosan [11] and alginate [2, 12, 13]) via physical reactions (e.g., change of temperature [11, 14, 15]), chemical cross-linking reactions with metal ions (e.g.,Ca2+) [2, 12, 13] and chemical crosslinkers [2, 16], photo-cross-linking reaction [10, 17–19], or enzymatic cross-linking reaction [20, 21]. Hydrogels comprised of naturally derived macromolecules have potential advantages of biocompatibility, biodegradation and intrinsic cellular interaction. Especially, hydrogels derived from naturally occurring macromolecules mimic features of extracellular matrix, have highly hydrated three-dimensional networks and potential to direct the migration, growth of cells during tissue regeneration and wound healing. They are also utilized for stabilization of encapsulated and transplanted cell [22] and sequester important growth factors that can augment tissue growth [23, 24].
Alginate is an attractive natural anionic linear polysaccharide composed of 1,4-linked β-d-mannuronate (MM-blocks) and 1,4-linked α-l-guluronate (GG-blocks) residues in variable proportions. Alginate has been widely used as wound dressing, drug delivery and cell encapsulation material, and injectable cell transplantation vehicle due to its biocompatibility, low toxicity and relatively low cost [22, 25–27]. An important feature of alginate is that it is freely soluble in water, and can be rapidly gelated in the presence of divalent cations, such as Ca2+ [12, 28].
In the previous reports, calcium sulphate [12] and calcium chloride [29] were usually used to form calcium alginate gels. However, calcium ions reacted with alginate rapidly, and calcium alginate gel is difficult to be formed homogeneously. So the bioinspired strategy of triggered release of calcium ions from thermally triggerable liposomes was developed successfully to induce gelation [13]. Hyun-Joon Kong et al. [30] incorporated NaCl and Na6(PO3)6 into the pre-gelled solutions to avoid an abrupt gelation process. In our experiments, in order to avoid the abrupt gelation process, trisodium phosphate was added into the pre-gelled solutions to develop a novel injectable system.
The bone-like nano-hydroxyapatite/collagen (nHAC) composites were synthesized biomimetically by mineralizing type I collagen, which possessed features similar to natural bone both in composition and hierarchical structure [31, 32]. The extensive researches on nHAC confirmed that it was biodegradable, biocompatible and bioactive [31–33]. But pure nHAC was difficult to mould. So porous nHAC/poly (L-lactic acid) composite and chitin fibre reinforced nHAC/poly (L-lactic acid) scaffold have been successfully developed in our lab [31–37]. These composites, however, are solid implants and need invasive surgical operations in clinical applications.
In this study, the calcium alginate hydrogel was expected as vehicle to carry nHAC particles to develop a novel injectable and in situ forming gel composite (GC) for bone-defect repair or bone tissue engineering. Trisodium phosphate was selected to add into the pre-gelled solution to manipulate the injectability of GC. The injectable, rheological, mechanical and in vitro degradable properties of GC were investigated. Meanwhile, the gelation mechanism of GC was discussed.
2 Materials and methods
2.1 Materials
Sodium alginate (W m = 2 ~ 3×105 g/mol) with 30–35% GG-blocks, trisodium phosphate (Na3PO4 · 12H2O) and calcium sulfate (CaSO4 · 2H2O) were purchased from Chemical Agents Co. Ltd, Beijing, China. The nHAC particles (50 ± 6.6 μm) were fabricated by mineralizing collagen using the method developed in our lab [31, 32]. The 0.1 mol/l phosphate buffered saline (PBS, pH = 7.4, Sigma) solution containing 1% penicillin/streptomycin (Sigma) was prepared for experiments. The colorimetric calcium reagent kit purchased from Sigma was used to test Ca2+ concentration. Alginate was sterilized by high-pressure steam sterilization [38], and the other materials were irradiated with a cobalt–60 source for 25 kGy.
2.2 Samples preparation
Sodium alginate, trisodium phosphate and nHAC were dissolved in deionized water to form hybrid solution, which is defined as solution A. In all experiments, nHAC was kept a constant concentration of 3% g/ml. Calcium sulfate was dissolved in deionized water, magnetically stirred for 1 h to form CaSO4 slurry, and then statically placed for more than 24 h. Before mixing, CaSO4 slurry needed to be stirred again for 1 min and then mixed together with solution A for 1 min. This pre-gelled solution was defined as solution B. The next step was putting solution B rapidly into the mould with a size of diameter × height = 2.0 cm × 1.6 cm to form cylindrical hydrogel for mechanical properties and in vitro degradation tests.
2.3 Injectable and rheological properties
The outflow volume and setting time are parameters to characterize the injectable properties. The 10 ml syringe (without needle) with 10 ml solution B was fixed vertically and imposed on a constant weight of 2 kg. The outflow volume in 1 min is the total volume (10 ml) minus surplus volume.
The rheological property of GC during the process of sol–gel transformation was tested by physica MCR300 Modular Compact Rheometer (Germany) with constant shear rate (\( \mathop \gamma \limits^{ \bullet } \, = \, 7. 3 4 {\text{s}}^{ - 1} \)). Because 3 min was needed to prepare solution B and put it into Rheometer, so the starting time of rheological test is the third min. Meanwhile, the initial setting time and the final setting time can be observed from the rheological curve.
2.4 pH value
The pH changing during the process of sol–gel transformation was tested via PHS-2F pH meter (Shanghai Precision Scientific Instrument Co., Ltd, China), and recorded at 1, 5, 10, 20, 30 and 60 min after solution A mixed with CaSO4 slurry, respectively.
2.5 X-ray diffraction
The GC samples were frozen in refrigerator at −20°C for 12 h and then lyophilized in freeze drier. XRD patterns of dry GC were obtained by XRD equipment, which recorded in a Rigaku D/max-RB diffractometer (Rigaku, Tokyo) with Cu anticathode.
2.6 FTIR spectroscopy
Infrared spectroscopy was used to characterize the structure of calcium alginate and GC. The infrared spectra of calcium alginate and GC were measured with an ATR-FTIR (Nicolet 560, US) spectrophotometer. Each spectrum of samples was acquired via accumulation of 256 scans with a resolution of 4 cm−1.
2.7 Mechanical test
Compressive elastic modulus (E) of GC was characterized by an universal testing machine (Model 1122, Instron Engineering Corp., Canton, MA) controlled with Test-Works 3.10 software (MTS Sintech Inc. Research Triangle Park, NC) as previously described [39–41]. The samples were compressed at a constant deformation rate of 1 mm/min. The stress and strain values obtained were adjusted by locating the 0 displacement, where the load changed from negative (due to surface tension effects) to positive (due to initial compressive loading) [12]. From the stress vs strain curves, the E value of GC was calculated. Since the curves tended to be non-linear, the strain range used for the calculation of the modulus was limited to the first 5% of the strain. Assuming that the GC fits to an affine network model, the shear modulus (G) was obtained from the slope σ vs –(λ–λ−2) plot, where σ is the stress, λ is the ratio of the deformed length to the undeformed length of GC sample [42, 43].
2.8 In vitro degradation
Approximately 3.6 g wet samples of GC (Group 4 in Table 2) was placed in 50 ml vial, each containing 20 ml 0.1 M PBS. The vials were incubated at 37°C in a shaking incubator with 40 rpm for various time periods. Each week the buffer solution was replaced by a fresh one. At the end of each time point, samples were removed from the solution, washed with distilled water and weighted. Then these samples were frozen in a refrigerator at −20°C for 12 h, and lyophilized, leaving the dry matrix for dry weight test. Finally, the corresponding wet and dry weights were recorded for analysis of the weight loss.
In addition, three wet samples were harvested to test the compressive strength and modulus at each experimental time point. The degradation solution was collected on day 1, 4, 7, 10, 14 and 21, and the pH values of the solution were tested by PHS-2F pH meter. Ca2+ ions concentration in degradation solution was determined by colorimetric calcium reagent kit. Readings were performed at 570 nm (Bio-Rad 680, USA). A calcium standard was used to prepare the calibration curve.
2.9 Statistical analysis
Three examples were used in each test and their average values were calculated for statistically analysis. All data were statistically analyzed by analysis of variance (ANOVA) software and expressed as mean ± standard deviation (SD). The difference was considered statistically significant when p < 0.05.
3 Results and discussion
3.1 XRD and FTIR analysis
The calcium sulphate, calcium phosphate and hydroxyapatite crystal phases existed in this injectable system can be proved by XRD analysis. The results were shown in Fig. 1. Previous researchs have confirmed hydroxyapatite crystal phases were the main inorganic phase of nHAC [32, 33], The XRD results indicated that the nHAC preserved its structural characteristics during gelation, which is favorable to maintain its bioactivity and biocompatibility.

XRD patterns of GC (a) and nHAC (b)
Comparing FTIR spectra of GC and alginate in Fig. 2, the characteristic peaks of alginate are –COO− (1,620 cm−1 and 1,410 cm−1) and –C–OH (O–H stretching vibration is 3,450 cm−1, C–O stretching vibration of secondary alcohol is 1,110 cm−1, C–O stretching vibration of tertiary alcohol is 1,150 cm−1), which verified that the chemical structure of alginate in GC was not changed. In addition, the hydroxyapatite characteristic peaks of 1,040 cm−1, 560 cm−1 and 600 cm−1 can be observed in spectra of GC, which also confirmed that nHAC maintained its structure in GC. According to the results shown in Figs. 1, 2, it can be concluded that nHAC and alginate maintained their structure in the process of GC preparation, which is beneficial to keep the biological characteristics of nHAC and alginate for bone repair.

FTIR spectra of nHAC (a), sodium alginate (b) and GC (c)
3.2 Injectable and rheological properties
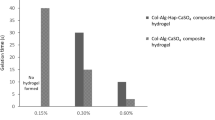
The rheological properties of the pre-gelled solution are critical to its applications [44]. The variation of rheological proprieties (e.g., viscosity and fluidity) was expected to be a character of injectability of GC. Increasing the concentration of alginate, the outflow volume decreased. Statistical analysis showed a significant effect of alginate concentration on the injectability (P < 0.05) (Table 1). The reason may be that the fluidity becomes weaker for the viscosity increased with alginate concentration increasing, as shown in Fig. 3a. Meanwhile, statistical analysis of the data also showed that increasing alginate concentration led to the process accelerating of sol–gel phase transformation as well as time from initial setting time to final setting time shortened significantly in Fig. 3b (P < 0.05). The initial setting time and the final setting time in the curves of Fig. 3b were from Fig. 3a. The time points were remarked as dashed (i and f) in curves in Fig. 3a. In addition, according to the initial setting time points and the final setting time points in Fig. 3a, three phases can be distinguished in the process of GC gelation: pre-gel, sol–gel phase transformation and post-gel (Fig. 3c). In pre-gel zone, the viscosity is the lowest in three phases, but the viscosity increased in the sol–gel phase transformation and its speed varied dramatically with the increasing of alginate concentration. Finally, the viscosity stabilized in post-gel zone. In addition, the gelling time increased with increasing of Na3PO4 · 12H2O (Table 2). In the range of all gelling times, surgeons suggested that 5–30 mins for gelation in a surgical environment was accepted. Analyzing the above results comprehensively, it is sure that injectability of GC system can be regulated by adjusting concentration of components. Considering the rheological properties of pre-gelled solution, mechanical properties (Fig. 6 and Table 2) and gelling time for GC, the system containing 5.0 mg/ml Na3PO4 · 12H2O and 3% sodium alginate (Group 4 in Table 2) has the optimal properties for future application in all GC systems.

a Rheological curve of GC with different alginate concentration. b The initial setting time (■) and the final setting time (●) curve of GC with different alginate concentration. c The rheological curve containing 3% alginate shows the three phases in the process of GC gelation
3.3 The pH value
As shown in Fig. 4, the pH values change rapidly from 11.8 to 7.6 during the process of GC gelation, and the pH values stabilize ultimately around 7.6 after 30 min. The pH value keeps invariance in the physiological pH range, which is a basic requirement for biomaterials. The pH values of GC were stabilized ultimately around 7.6 in a short time, which meets the need of biomaterials for applications.

The pH change during the process of sol–gel transformation
3.4 Mechanical properties
The mechanical properties of the gelled material in the body (e.g., rigidity and brittleness) are important parameters to maintain the gel structure in the face of the compression from neighboring tissues [45]. In addition, the mechanical stiffness can potentially be utilized to regulate the gene expression of the cells distributed in hydrogels [46]. Therefore, it is highly necessary to attain mechanical properties of GC. Samples for mechanical properties test were shown in Fig. 5. Here, it was mainly tested that the influences of concentrations of trisodium phosphate and sodium alginate on mechanical properties (Fig. 6 and Table 2). The E value increased at first, and then decreased with the increasing trisodium phosphate content (Table 2). Once trisodium phosphate is relatively deficient (Group 1–3 in Table 2), the rapid gelling of GC resulted in inhomogenous and serious defects in the gels, which led to weaker mechanical strength than that of group 4. The mechanical strength in group 5–7 is also weaker than that of group 4. It may be caused by more trisodium phosphate reacted with calcium sulfate, which lead to calcium sulfate relatively shortage. As a result, less calcium ions could participate in cross-linking alginate, and the cross-linking density decline. As seen in Fig. 6, the variations of E and G values are not linear with the increasing alginate concentration. It is considered that the molecules intertwisted mutually and cross-linking density increased with the increasing of alginate concentration, which led to the general tendency of mechanical strength ascending, but nHAC and Ca3(PO4)2 particles existed in gel may result in stress concentration and decreasing of mechanical strength.

Samples for mechanical property and in vitro degradation tests

Compressive modulus E (a) and shear modulus G (b) for GC with different alginate concentrations
3.5 In vitro degradation
In this in vitro degradation experiment, the wet weight increased in the first week, and it shows the negative value in wet weight loss curve (Fig. 7a), but dry weight loss increased at all times (Fig. 7b). This result indicates that samples absorbed water after immerged into PBS solution. The absorbed water is heavier than the weight loss, which led to the wet weight increase in the first week. Statistical analysis of the degradation data after 4 weeks showed that the wet weight and dry weight loss reached to 42.5% and 49.7%, respectively. Accompanying decreasing weight, E and G values declined at all time point (Table 3). After 2 weeks, the E and G values could not be tested for surface change, but no samples broke down up to 28 days. The pH values of degradation solution had no obvious change during in vitro degradation with no statistically significant differences (P < 0.05) (Fig. 8). The pH values kept in the physiological pH range that would be benefit to material biocompatibility. Ca2+ ions concentration also had no significant change during in vitro degradation without statistically significant differences (P < 0.05). There was no burst of calcium release (Fig. 9). The main reason was that Ca3(PO4)2, CaSO4 and Ca-alginate could not produce large number of free Ca2+ in GC system, because they are sparingly soluble or insoluble salts with small solubility product constant: K sp (CaSO4) ≫ K sp (Ca-alginate) > K sp (Ca3(PO4)2) (detail discussion in section 3.6).

Wet weight loss (a) and dry weight loss (b) of GC during in vitro degradation

The pH change during in vitro degradation

Calcium release from GC system
Degradability is an important parameter of materials for many applications in tissue engineering [47], the degradation of alginate hydrogels is very slow and poorly controlled. To resolve this problem, Bouhadir [48] reported an approach to control alginate gel degradation via partial periodate oxidation. This approach offered control over the degradation rate by varying the degree of oxidation. However, the biocompatibility of alginate is likely to decrease at high degree of oxidation for the presence of aldehyde groups on the sugar residues. Meanwhile, mechanical strength declines significantly. During the in vitro degradation of GC, the wet weight and dry weight lost gradually, and surface changed greatly in 2 weeks, but no samples broke down up to 28 days. From this phenomenon, it could be inferred that GC degraded from outside to inside.
3.6 Gelation mechanism of GC
To understand the mechanism of the process of sol–gel transformation, we try to discuss the reasons for this phenomenon. The formation process of the composite is inferred as follows:
Since CaSO4 is a sparingly soluble salt, it can only produce a small quantity of Ca2+ in aqueous environment (Eq. 1). In GC system, the Ca2+ from CaSO4 reacted with PO4 3− firstly (Eq. 2), and produced insoluble calcium phosphate because the solubility product constant of Ca3(PO4)2 (K sp (Ca3(PO4)2) = 2.07 × 10−29, 298.15 K) is much lower than that of CaSO4 (K sp (CaSO4) = 4.93 × 10−5, 298.15 K). Then the surplus CaSO4 unceasingly produced Ca2+, which cross-linked carboxyls of alginate to form calcium alginate hydrogel (Eq. 3). Calcium alginate hydrogel (Ca-alginate) formed via Ca2+ released from CaSO4, meaning that the stability of CaSO4 is weaker than that of Ca-alginate, as well as K sp (CaSO4) > K sp (Ca-alginate) in spite of K sp (Ca-alginate) can not be tested exactly. Here, therefore, the stability of Ca-alginate, CaSO4 and Ca3(PO4)2 can be inferred as follows: CaSO4 < Ca-alginate < Ca3(PO4)2, as a result of K sp (CaSO4) ≫ K sp (Ca-alginate) > K sp (Ca3(PO4)2).
In order to validate above hypothetical process, trisodium phosphate was added into the pre-gelled solutions with different concentrations. As more than 8.0 mg/ml Na3PO4 · 12H2O was dissolved into 40 ml containing 3% alginate solution, and reacted with 5 ml slurry of calcium sulfate, no gelation occurred after more than 24 h and a lot of deposition appeared (Group 7 in Table 2), which confirmed that Ca2+ reacted with PO4 3− ions firstly in process of GC gelation, or else Ca2+ reacted with alginate rapidly to form gel. In addition, the XRD results of GC (Fig. 1) showed that calcium phosphate and calcium sulfate are present, providing another evidence for the above gelation mechanism. Two reasons may lead to the variation of the setting time. One is the process of calcium sulfate translating into the insoluble calcium phosphate, which can prolong the gelling time. The other important reason is that Ca3(PO4)2 deposited on CaSO4 particles to hinder dissolving of CaSO4 and producing of Ca2+.
4 Conclusion
GC is an injectable and in situ-forming biomaterial consisting of calcium alginate hydrogel and nHAC. The injectability of GC system can be tunable by adjusting concentration of components. According to the rheological curves, there are three phases of pre-gel, sol–gel phase transformation and post-gel existed in the process of GC gelation. The results of XRD and FTIR confirmed that the alginate and nHAC can maintain their own structure characters. The pH values of GC can be stabilized ultimately around 7.6 in 30 min, which satisfied the requirement for biomaterials. The E and G values are in the range of 17.0–56.0 kPa and 24.7–55.0 kPa, respectively. In the in vitro degradation test, the wet weight increased in the first week for samples absorbed water after immerged in PBS. After 4 weeks in vitro degradation, wet weight loss and dry weight loss reached to 42.5% and 49.7%, respectively. Meanwhile, the samples did not break down up to 28 days. In a word, GC has the controllable setting time and tunable injectability, which provides a possible injectable material for bone repair and bone tissue engineering.
References
W. Bensaid, J.T. Triffitt, C. Blanchat, K. Oudina, L. Sedel, H. Petite, Biomaterials 24, 2497 (2003). doi:10.1016/S0142-9612(02)00618-X
B. Balakrishnan, A. Jayakrishnan, Biomaterials 26, 3941 (2005). doi:10.1016/j.biomaterials.2004.10.005
K.Y. Lee, D.J. Mooney, Chem. Rev 101, 1869 (2001). doi:10.1021/cr000108x
S.K. Mallapragada, B. Narasimhan, Biomaterials 23, 4305 (2002). doi:10.1016/S0142-9612(02)00172-2
B. Jeong, Y.H. Bae, D.S. Lee, S.W. Kim, Nature 388, 860 (1997). doi:10.1038/42218
B.Ф. Palsson, S.N. Bhatia, Tissue Engineering (Person Prentice Hall, USA, 2004), pp. 261–262. Person Eduction,Inc
M.D. Timmer, C.G. Ambrose, A.G. Mikos, Biomaterials 24, 571 (2003). doi:10.1016/S0142-9612(02)00368-X
R.G. Payne, M.J. Yaszemski, A.W. Yasko, A.G. Mikos, Biomaterials 23, 4359 (2002). doi:10.1016/S0142-9612(02)00184-9
J.P. Fisher, T.A. Holland, D. Dean, P.S. Engel, A.G. Mikos, J. Biomater. Sci. Polym. Ed 12, 673 (2001). doi:10.1163/156856201316883476
J.P. Fisher, D. Dean, A.G. Mikos, Biomaterials 23, 4333 (2002). doi:10.1016/S0142-9612(02)00178-3
A. Chenite, C. Chaput, D. Wang, C. Combes, M.D. Buschmann, C.D. Hoemann, J.C. Leroux, B.L. Atkinson, F. Binette, A. Selmani, Biomaterials 21, 2155 (2000). doi:10.1016/S0142-9612(00)00116-2
T. Boontheekul, H.J. Kong, D.J. Mooney, Biomaterials 26, 2455 (2005). doi:10.1016/j.biomaterials.2004.06.044
E. Westhaus, P.B. Messersmith, Biomaterials 22, 453 (2001). doi:10.1016/S0142-9612(00)00200-3
K.E. Crompton, J.D. Goud, R.V. Bellamkonda, T.R. Gengenbach, D.I. Finkelstein, M.K. Horne, J.S. Forsythe, Biomaterials 28, 441 (2007). doi:10.1016/j.biomaterials.2006.08.044
J.H. Cho, S.H. Kim, K.D. Park, M.C. Jung, W.I. Yang, S.W. Han, J.Y. Noh, J.W. Lee, Biomaterials 25, 5743 (2004). doi:10.1016/j.biomaterials.2004.01.051
K. Nam, T. Kimura, A. Kishida, Biomaterials 28, 1 (2007). doi:10.1016/j.biomaterials.2006.08.002
S. Seiffert, W. Oppermann, K. Saalwaechter, Polymer 48, 5599 (2007). doi:10.1016/j.polymer.2007.07.013
G. Pitarresi, M.A. Casadei, D. Mandracchia, P. Paolicelli, F.S. Palumbo, G. Giammona, J. Control Release 119, 328 (2007). doi:10.1016/j.jconrel.2007.03.005
G. Pitarresi, F.S. Palumbo, G. Giammona, M.A. Casadei, F.M. Moracci, Biomaterials 24, 4301 (2003). doi:10.1016/S0142-9612(03)00332-6
V. Crescenzi, A. Francescangeli, A. Taglienti, Biomacromolecules 3, 1384 (2002). doi:10.1021/bm025657m
T.H. Chen, H.D. Embree, E.M. Brown, M.M. Taylor, G.F. Payne, Biomaterials 24, 2831 (2003). doi:10.1016/S0142-9612(03)00096-6
G.D. Nicodemus, S.J. Bryant, Tissue Eng. Pt. B-Rev 14, 149 (2008). doi:10.1089/ten.teb.2007.0332
X.Z. Shu, S. Ahmad, Y.C. Liu, G.D. Prestwich, J. Biomed. Mater. Res. A 79A, 902 (2006). doi:10.1002/jbm.a.30831
S.S. Cai, Y.C. Liu, X.Z. Shu, G.D. Prestwich, Biomaterials 26, 6054 (2005). doi:10.1016/j.biomaterials.2005.03.012
H.H. Tonnesen, J. Karlsen, Drug Dev. Ind. Pharm 28, 621 (2002). doi:10.1081/DDC-120003853
C.Y. Yu, X.C. Zhang, F.Z. Zhou, X.Z. Zhang, S.X. Cheng, R.X. Zhuo, Int. J. Pharm 357, 15 (2008). doi:10.1016/j.ijpharm.2008.01.030
A. Kikuchi, T. Okano, Adv. Drug Deliv. Rev 54, 53 (2002). doi:10.1016/S0169-409X(01)00243-5
B.L. Strand, Y.A. Morch, G. Skjak-Braek, Minerva Biotechnol 12, 223 (2000)
L. Wang, R.M. Shelton, P.R. Cooper, M. Lawson, J.T. Triffitt, J.E. Barralet, Biomaterials 24, 3475 (2003). doi:10.1016/S0142-9612(03)00167-4
H.J. Kong, K.Y. Lee, D.J. Mooney, Polymer 43, 6239 (2002). doi:10.1016/S0032-3861(02)00559-1
C. Du, F.Z. Cui, W. Zhang, Q.L. Feng, X.D. Zhu, K. de Groot, J. Biomed. Mater. Res 50, 518 (2000). doi:10.1002/(SICI)1097-4636(20000615)50:4<518::AID-JBM7>3.0.CO;2-W
R.Z. Wang, F.Z. Cui, H.B. Lu, H.B. Wen, C.L. Ma, H.D. Li, J. Mater. Sci. Lett 14, 490 (1995). doi:10.1007/BF00665911
S.S. Liao, F.Z. Cui, W. Zhang, Q.L. Feng, J. Biomed. Mater. Res. B 69B, 158 (2004). doi:10.1002/jbm.b.20035
S.S. Liao, F.Z. Cui, Y. Zhu, J. Bioact. Compat. Polym 19, 117 (2004). doi:10.1177/0883911504042643
X.M. Li, Q.L. Feng, X.H. Liu, W. Dong, F.H. Cui, Biomaterials 27, 1917 (2006). doi:10.1016/j.biomaterials.2005.11.013
X.M. Li, Q.L. Feng, F.Z. Cui, Mater. Sci. Eng. C-Bio 716, 26 (2006)
X.M. Li, Q.L. Feng, Y.F. Jiao, F.H. Cui, Polym. Int 54, 1034 (2005). doi:10.1002/pi.1804
K.T. Paige, L.G. Cima, M.J. Yaremchuk, J.P. Vacanti, C.A. Vacanti, Plast. Reconstr. Surg 96, 1390 (1995)
Q. Lv, Q.L. Feng, J. Mater. Sci.: Mater. Med 17, 1349 (2006). doi:10.1007/s10856-006-0610-z
K.S. Anseth, C.N. Bowman, L. BrannonPeppas, Biomaterials 17, 1647 (1996). doi:10.1016/0142-9612(96)87644-7
X.M. Li, Q.L. Feng, Mater. Sci. Forum 475–479, 2387 (2005)
R. Skouri, F. Schosseler, J.P. Munch, S.J. Candau, Macromolecules 28, 197 (1995). doi:10.1021/ma00105a026
E.C. Muniz, G. Geuskens, Macromolecules 34, 4480 (2001). doi:10.1021/ma001192l
J.S. Temenoff, A.G. Mikos, Biomaterials 21, 2405 (2000). doi:10.1016/S0142-9612(00)00108-3
B.S. Kim, D.J. Mooney, Trends Biotechnol 16, 224 (1998). doi:10.1016/S0167-7799(98)01191-3
D. Ingber, S. Karp, G. Plopper, L. Hansen, D.J. Mooney, Physical forces and the mammalian cell (Academic Press, New York, 1993)
LC Lu, X Zhu, RG Valenzuela, BL Currier, MJ Yaszemski, Clin. Orthop. Relat. R. S251 (2001)
K.H. Bouhadir, K.Y. Lee, E. Alsberg, K.L. Damm, K.W. Anderson, D.J. Mooney, Biotechnol. Prog 17, 945 (2001). doi:10.1021/bp010070p
Acknowledgments
The authors are grateful for the financial support from National Great Project of Science and Technology Supporting Programs of China during the 11th Five-year Plan (No. 2006BAI16B03) and National Natural Science Foundation of China (No. 50803032).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, R., Niu, X., Gan, S. et al. Preparation and characterization of an injectable composite. J Mater Sci: Mater Med 20, 1245–1253 (2009). https://doi.org/10.1007/s10856-009-3692-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-009-3692-6