
Abstract
Hydrogen energy is regarded as one of the most important clean energy in the twenty-first century, and improving the catalytic efficiency of hydrogen evolution reaction (HER) is the basis for realizing the large-scale hydrogen production. Transition metal phosphides (TMPs) were proved to be efficient electrocatalysts for HER. In this work, we first synthesized the nickel-molybdenum bimetallic precursors, followed by high-temperature calcination in air. Finally, NiMoP/MoP nanorods (Ni–Mo–P NRs) was obtained by chemical vapor deposition (CVD) of phosphating. The target catalyst of Ni–Mo–P NRs was characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). For Ni–Mo–P NRs, the electrochemical test in 0.5 M H2SO4 solution for HER showed that the optimal feeding ratio was Ni:Mo = 1:1. And the Ni1–Mo1–P NRs presented an onset potential of 63.2 mV, and an overpotential of 117.9 mV was required to drive the current density of 10 mA cm−2. Meanwhile, The Tafel slope, exchange current density (j0), electrochemical double-layer capacitance (Cdl) were 58.6 mV dec−1, 0.10 mA cm−2, 12.6 mF cm−2, respectively. Moreover, there was no obvious activity diminish of Ni1–Mo1–P NRs after a long-term stability and durability test.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the past few years, as the society developed rapidly and the fast consumption of traditional fossil fuels, the energy crisis and ecological environment problems caused by them have attracted great attention, which forces people to seek an ideal renewable energy source [1,2,3,4]. As a desirable clean energy, energy density of hydrogen can reach up to 120–140 MJ kg−1 [5]. And HER is a half reaction of water electrolysis, which has no pollution to the environment. However, in view of the low efficiency of HER process and the large overpotential caused by slow kinetic processes, an efficient catalyst capable of overcoming the energy barrier is needed to accelerate the reaction rate [6,7,8]. Commercial Pt-based catalysts are recognized as the catalysts with best performance so far, but their high price and instability in strong acid or alkaline medium limit the industrialization of hydrogen production [9, 10]. Inexpensive, high-efficiency and steady-going electrocatalysts need to be explored urgently.
Ideal electrocatalysts should not only have more exposed active sites and excellent conductivity, but also have special surface structures that facilitate rapid removal of hydrogen (H2), such as nanorods, nano-polyhedrons, nanotubes [11,12,13]. In addition, researchers have currently explored a range of transition metal catalysts, for example, carbides (TMCs), nitrides (TMNs), sulfides (TMSs), phosphides (TMPs) for HER. Among them, TMPs like FeP [14, 15], Ni2P [16, 17], CoP [18, 19], were deemed to be promising electrocatalysts since its catalytic mechanism is analogous to that of hydrogenase. In addition, P in TMPs can be used as a proton receptor center to promote the formation of transition metal hydrides, thus promoting the subsequent electrochemical desorption process. Furthermore, TMPs have better stability under extreme conditions.
Lately, molybdenum (Mo) phosphides were expected to replace precious metals catalysts for hydrogen evolution reaction because of their low-cost, high activity and strong durability [20,21,22]. Yang et al. used the MOF to synthesize MoP@PC showed excellent catalytic activity toward HER. The overpotential of 153 mV were needed to drive a current density of 10 mA cm−2 and the Tafel slope was 66 mV dec−1 [23]. Besides, Ni2P was regarded as a highly active catalyst since it was familiar with the catalytic mechanism of ferronickel hydrogenase and has been widely studied among TMPs in recent years [24, 25]. In addition, studies have shown that bimetallic TMPs such as Fe–Co–P [26, 27], Fe–Ni–P [28,29,30], Ni–Co–P [31,32,33] exhibited good HER or OER activity due to the synergistic effect between bimetals, which enhanced the conductivity, optimized the reaction adsorption energy and reduced the reaction energy barrier in the catalytic process. Moreover, the reasonable design of hybrid architecture is also a significant method to optimize HER activity [34].
Based on the analysis above, we designed and synthesized heterogeneous Nix–Moy–P NRs (x, y represented the molar ratio of Ni to Mo) by a simple and efficient three-step method. Electrochemical measurements under acidic conditions for HER showed that Ni1–Mo1–P NRs had the best activity with an onset potential (η0) of 63.2 mV, Tafel slope of 58.6 mV dec−1, an overpotential of 117.9 mV at 10 mA cm−2, respectively. Furthermore, Ni1–Mo1–P NRs maintained the activity and morphology well even after long-term stability test.
2 Experimental
2.1 Materials
Nickel chloride hexahydrate (NiCl2·6H2O, AR), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, AR), urea (CO(NH2)2, AR) and diammonium hydrogen phosphate ((NH4)2HPO4, AR) were all purchased from Aladdin Industrial Corporation (Shanghai, China). All chemical reagents were used without further purification.
2.2 Preparation of catalysts
2.2.1 Preparation of NixMoy precursor
Preparation of Ni1Mo1 precursor referred to the literature with slight modification [35]. Solution A: NiCl2·6H2O (1 mmol, 0.238 g) and urea (5 mmol, 0.3 g) dissolved in 30 mL deionized water and stirred vigorously; Solution B: (NH4)2Mo7O24·6H2O (1 mmol, 1.235 g) was completely dissolved in 30 mL deionized water; Solution C: solution B was added to solution A dropwise under strong magnetic stirring. The solution C was transferred to a 100 mL Teflon-lined stainless-steel autoclave and reacted at 160 °C for 16 h. After cooling down to ambient temperature, the product was collected by centrifugation and washed with deionized water and absolute ethanol for several times, and then dried at 60 °C in oven for 12 h. The control experiment adjusted the molar ratio of Ni to Mo (x:y = 1:0, 1:2, 2:1) under the same condition.
2.2.2 Preparation of Nix–Moy–O NRs
Put an appropriate amount of NixMoy precursor in a porcelain boat and subjected to 400 °C treatment under air for 3 h under air with a temperature ramping rate of 5 °C min−1. Take it out after cooling down to room temperature for further use.
2.2.3 Preparation of Nix–Moy–P NRs
(NH4)2HPO4 as phosphorus source and Nix–Moy–O NRs were placed separately in a porcelain boat followed by introducing 10% H2/Ar, then heated to 850 °C and maintained for 2 h with a temperature ramping rate of 5 °C min−1. After the reaction, the catalyst was cooled to room temperature and taken out for electrochemical performance testing.
2.3 Characterization
The surface morphology, structure and chemical composition of the samples were characterized by TEM (JEM-1400F, JEOL, Japan) with energy dispersive X-ray spectrum (EDS), XRD (Rigaku MiniFlex-600) with a Cu radiation source (λ = 0.1540 nm), SEM (SU8020, Hitachi, Japan), XPS ((ESCALAB250Xi, Thermo, United States), respectively.
2.4 Electrochemical test
All electrochemical tests were performed in a three-electrode system at room temperature on an electrochemical workstation of CHI760 with an electrolyte of 0.5 M H2SO4 solution saturated with N2. The glassy carbon electrode (GCE, area is 0.196 cm2) was used as the working electrode, while the Ag/AgCl electrode was used as the reference electrode and the carbon rod was used as the counter electrode. Firstly, 5 mg of the prepared catalyst was evenly dispersed in 1 mL of mixed solvent of absolute ethanol and Nafion (volume ratio: 19:1) by ultrasonicated for 0.5 h. Then a proper amount of well-dispersed catalyst solution was dropwise added onto the glassy carbon electrode with a pipette, and the test was conducted after natural drying. The scanning rate for the linear sweep voltammetry (LSV) test was 5 mV s−1. The cyclic voltammetry (CV) scan rate for stability and durability was 50 mV s−1. Electrochemical impedance spectroscopy (EIS) measurement was carried out with a frequency range from 100 kHz to 0.1 Hz at an amplitude of 5 mV. All potentials were corrected for standard hydrogen electrode potential according to the equation E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 + 0.059pH.
3 Results and discussion
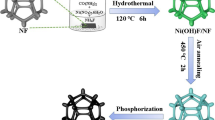
In this work, firstly, ammonium molybdate solution was added dropwise into the solution of nickel nitrate and urea to synthesize precursors of NixMoy bimetallic nanorods with different molar ratios of NiCl2·6H2O to (NH4)6Mo7O24·4H2O through simple hydrothermal reaction, and the color of the precursor gradually became lighter with the increase of Mo content. Then, the precursors were calcined into Nix–Moy–O NRs at high temperature in air. Finally, Nix–Moy–P NRs was obtained by CVD phosphating process. The detailed synthesis process was shown in Fig. 1.

Schematic illustration of synthesis procedure for Nix–Moy–P hierarchical nanorods
It can be found that in the absence of ammonium molybdate, the synthesized nickel precursor was a very thin nanowire, and its morphology was usually difficult to maintain at high temperature (Figs. S1b, S3a). The nano-particles of NiO (PDF NO.41-1049) were formed by high-temperature calcination (Figs. S4, S5b). When Ni:Mo ratio reached 1:1 and 2:1, the morphology of the as-prepared NixMoy are nanorods with smooth surface, but the size reached about 200 nm (Figs. S1a, d, S3b, d), and the XRD patterns are almost identical (Fig.S2). After pyrolysis in air, the XRD peaks at 14.3°, 24.0°, 29.8°, 33.3° and 44.1° were consistent with NiMoO4 (PDF No. 86-0361) (Fig. S4), and Figure S5a,d indicated good morphology. It should be noted that when the NiMo ratio was 1:2, black particles (Fig. S1c) emerged on the surface of the Ni1Mo2 precursor and it was likely that the excessive Mo species caused slight changes in the crystal structure and morphology. Besides, the diffraction peaks of MoO3 (PDF NO. 05-0508) appeared in XRD pattern after pyrolysis in addition to NiMoO4 (Fig. S4), and the morphology remained good after high-temperature calcination (Fig. S5c).
After Nix–Moy–O was phosphated by CVD method, Ni2P (PDF NO. 03-0953) nanoparticles was obtained when the molar ratio of Ni to Mo was 1:0 (Figs. 2, 3a). NiMo bimetallic oxide was converted into Nix–Moy–P dendritic nanorods (Fig. 3b–d), and the diffraction peaks at 30.5°, 35.3°, 39.2°, 47.3°, 49.2°, 58.9° correspond to the (110), (200), (111), (210), (002), (112) planes of NiMoP (PDF NO. 31-0873), while diffraction peaks at 27.9°, 32.2°, 43.1°, 57.5°, 58.0° and 67.9° perfectly corresponded to the (001), (100), (101), (110), (002) and (102) planes of MoP (PDF NO. 24-0771) (Fig. 2). With the increasing of Mo content, the peaks intensity of MoP strengthened while the peaks of NiMoP decreased gradually. Additionally, SEM images of Nix–Moy–P NRs (Fig. 4a–d) indicated that the rod-like structure were well maintained. As revealed by high-resolution TEM (HRTEM) images (Fig. 3e) of Ni1–Mo1–P NRs, the lattice fringes with interplanar spacing of 0.255 nm belonged to the (200) facet of MoNiP while the characteristic lattice spacing of 0.159 nm was ascribed to (002) of MoP. Besides, it can be known that the elements of Ni, Mo and P were distributed uniformly on the surface of Ni1–Mo1–P NRs (Fig. 3f), and corresponding atomic ratio was 40.27:30.94:28.80 (Table 1). The optimum ratio of nickel and molybdenum metal atoms facilitates crystal growth in a particular lattice plane. Table S1 listed the atomic percentage of Ni, Mo and P in Nix–Moy–P NRs.

XRD patterns of a Nix–Moy–P NRs

TEM images a–d of Ni1–Mo0–P NPs, Ni1–Mo1–P, Ni1–Mo2–P, Ni2–Mo1–P NRs, e high-resolution TEM images of the Ni1–Mo1–P NRs, f TEM image of Ni1–Mo1–P NRs and corresponding elemental mapping images

SEM images of a Ni1–Mo1–P NPs, b Ni1–Mo2–P NRs, c Ni2–Mo1–P NRs, d Ni1–Mo0–P NPs, e EDS spectra of Ni1–Mo1–P NRs
To further determine the elemental content and valence state of Ni1–Mo1–P NRs, XPS measurement was executed. The existence of Ni, Mo, P, O was confirmed by the XPS full spectrum of Ni1–Mo1–P NRs (Fig. 5a), which was consistent with EDS results (Fig. 4e). In the high-resolution XPS spectra of Mo 3d (Fig. 5b), the two peaks at the binding energies (BE) of 227.8 eV and 231.1 eV can be attributed to the metal phosphide Mo-P, and the doublets at 232.48 eV and 235.38 eV corresponded to the oxidation state of Mo due to the higher surface energy of MoP [36,37,38]. In the region of Ni 2p (Fig. 5c), it should be noted that the peaks at 853.1 eV and 873.9 eV could be assigned to Ni 2p3/2 and Ni 2p1/2 in Ni–P which had a positive shift compared with that of Ni1–Mo0–P NPs because of the electronegativity difference between Ni (1.91) and Mo (2.16) [17, 22]. The doublets at 857.1 eV and 875.33 eV could be ascribed to the oxidation state of Ni species because of passivation in air while two peaks at 862.4 eV and 880.8 eV were satellite peaks [39, 40]. For P 2p spectra (Fig. 5d), characteristic peaks located at 129.3 eV and 130.3 eV were designated to metal phosphide (P 2p3/2 and 2p1/2) [18], which showed mildly negative offset compared to P 2p in Ni1–Mo0–P NPs. Furthermore, the broad peak near 134.2 eV was attributed to oxidized P substances such as phosphates and phosphorus pentoxide [9, 15]. It is observed that nickel has a partial positive charge (δ+) while phosphorus had a partial negative charge (δ−), which indicated that the electron was transferred from metal element to phosphorus element. These phenomena mean that the addition of Mo result in the charge redistribution in catalysts and reduce the energy barrier of the electrocatalytic process, which promoted the adsorption and dissociation process of water in HER [41, 42].

XPS spectra of the Ni1–Mo1–P NRs and Ni1–Mo0–P NPs a survey, b Mo 3d, c Ni 2p, d P 2p
The electrocatalytic properties of the Ni1–Mo1–P NRs towards HER was investigated by the linear sweep voltammetry (LSV) using a standard three-electrode electrochemical cell in 0.5 M H2SO4 solution saturated by N2. The control samples of Ni2–Mo1–P NRs, Ni1–Mo2–P NRs, Ni1–Mo0–P NPs electrodes were also studied under the same conditions for comparison. As shown in LSV curves (Fig. 6a), the Ni1–Mo1–P NRs exhibited promising HER performance with an ultrasmall onset potential of 63.4 mV and an overpotential of 117.9 mV at a current density of 10 mA cm−2, which was much lower than those of Ni2–Mo1–P NRs (170.3 mV), Ni1–Mo2–P NRs (228.8 mV), and Ni1–Mo0–P NPs (329.1 mV), suggesting that Ni1–Mo1–P NRs had high activity toward HER in acidic electrolyte. To display the outstanding catalytic activity of Nix–Moy–P NRs, the required overpotentials of as-prepared catalysts at different current density of 1, 10, 20 mA cm−2 were presented in Fig. 6b.

a LSV curves, b η1, η10 and η20, c corresponding tafel slopes, d EIS Nyquist plots; e Cdl at 0.26 V as a function of scan rates and f j0 of different catalysts in 0.5 M H2SO4 solution
The Tafel slope was calculated from the LSV curve based on Tafel equation: η = b lg j + a (b, j represented Tafel slope and current density), and comparisons of Tafel slopes were demonstrated in Fig. 6c. The Tafel slope of Ni1–Mo1–P NRs was 58.6 mV dec−1, which was smaller than that of Ni2–Mo1–P (70.7 mV dec−1), Ni1-Mo2-P (91.7 mV dec−1) and Ni1–Mo0–P (147.2 mV dec−1), indicating faster kinetics of Ni1-Mo1-P NRs in HER. Typically, H2 production involves two different reaction pathways, namely, Vollmer–Heyrovsky and Volmer–Tafel mechanisms, as determined by the change of Tafel slope [43]. Theoretically, the Tafel slopes corresponding to Volmer, Heyrovsky and Tafel processes are 120, 40 and 30 mV dec−1, respectively [44]. Ni1–Mo1–P NRs manifested the Tafel slopes of 58.6 mV dec−1, suggesting a Volmer–Heyrovsky mechanism for Ni1–Mo1–P NRs and the Heyrovsky process was the rate-limiting step.
Furthermore, the electrochemical impedance spectroscopy (EIS) was measured to further explore the HER kinetics of Ni1–Mo1–P NRs. The equivalent circuit diagram embedded in Fig. 6d, where Rs, CPE, Rct stands for the solution resistance, constant-phase element, and charge-transfer resistance, respectively [45, 46]. The semicircle diameter represents the charge transfer resistances (Rct), which can evaluate the charge transfer capability. The measured Rct of Ni1–Mo1–P NRs was 28.4 Ω was lower than Ni2–Mo1–P NRs (33.8 Ω), Ni1–Mo2–P NRs (60.9 Ω) and Ni1–Mo0–P NPs (72.2 Ω), owing to its ample synergetic interfaces that benefits the integration of electrons and reactants at the catalyst-electrolyte interphase and short diffusion [47, 48].
To investigate the intrinsic activity of materials, the electrochemical surface areas (ECSA) of the catalysts were also studied by testing the double layer capacitance (Cdl) at varied scan rates. The CV curves for Ni1–Mo1–P NRs, Ni2–Mo1–P NRs, Ni1–Mo2–P NRs and Ni1–Mo0–P NPs (Fig. S6) within 0.05–0.45 V at different scanning rates of 10–100 mV s−1 were performed. The Cdl was measured by plotting the difference between anode current (ja) and cathode current (jc) at a constant potential 0.26 V vs reversible hydrogen electrode (RHE) with respect to the scanning rates. As shown in Fig. 6e and Fig. S7, Ni1–Mo1–P NRs had the highest Cdl (12.6 mF cm−2) and ECSA (315 cm2) among all catalysts, which indicated that more active sites were exposed on the surface of Ni1–Mo1–P NRs to improve HER performance [49, 50].
The exchange current density (j0) is another important criterion for evaluating HER activity which can be obtained by extrapolation from Tafel slope diagram (Fig. 6f). The j0 for Ni1–Mo1–P NRs was 0.10 mA cm−2, which was higher than other catalysts, further proving the excellent performance. Table 2 listed the performance comparison of several catalysts under acidic conditions in recent years.
In addition to the excellent catalytic performance, stability is an essential criterion for judging the quality of catalysts. Stability and durability test of Ni1–Mo1–P NRs were performed in 0.5 M H2SO4 solution as shown in Fig. 7a. The current density decayed by 5.4 mV at a current density of 10 mA cm−2 after continuously scanning for 2000 times using cyclic voltammetry. Meanwhile, the durability of Ni1–Mo1–P NRs was determined by a chronoamperometric curve, and the catalytic activity was maintained at 82.8% after 12 h of continuous testing. This indicated that Ni1–Mo1–P NRs has good stability and corrosion resistance. Moreover, according to the comparison of XRD patterns before and after the stability test (Fig. 7b), it was quite clear that the crystal structure of Ni1–Mo1–P NRs was almost unchanged, which also indicated better stability and durability of the target catalyst.

a stability test of Ni1–Mo1–P NRs in 0.5 M H2SO4 solution, b Comparison of XRD patterns before and after electrolysis,
Excellent HER activity and stability can be attributed to the following aspects: (1) bimetallic phosphides can effectively improve the adsorption of hydrogen, and the incorporation of Mo can effectively change the charge distribution of Nix–Moy–P NRs; (2) Ni1–Mo1–P NRs has good conductivity, faster charge transfer kinetics and more active sites; (3) One-dimensional structure also contributes to excellent durability and long-term stability.
4 Conclusions
To sum up, Ni1–Mo1–P NRs with high performance for HER was prepared by simple hydrothermal method, high temperature calcination and CVD method in turn. The results showed that the addition of Mo optimized the electronic distribution of catalyst, and Ni1–Mo1–P NRs represented excellent HER activity with a small onset potential of 63.4 mV and super stability in acidic medium. This work provides a feasible strategy for preparing other transition metal phosphorus heterostructure electrocatalysts, which can realize steady and robust electrochemical hydrogen production. The abundant metal source, highly efficient and stable catalytic activity make Ni1–Mo1–P NRs become one of the potential catalysts for hydrogen production industrialization.
References
C.G. Morales-Guio, L.A. Stern, X. Hu, Chem. Soc. Rev. 43, 6555–6569 (2014)
B. Hua, M. Li, Y.F. Sun, Y.Q. Zhang, N. Yan, J. Chen, T. Thundat, J. Li, J.L. Luo, Nano Energy 32, 247–254 (2017)
H. Wu, X. Lu, G. Zheng, G.W. Ho, Adv. Energy Mater. 8, 1702704 (2018)
X. Sun, J. Huo, Y. Yang, L. Xu, S. Wang, J. Energy Chem. 26, 1136–1139 (2017)
Y. Yan, B.Y. Xia, B. Zhao, X. Wang, J. Mater. Chem. A 4, 17587–17603 (2016)
Y. Zheng, Y. Jiao, M. Jaroniec, S.Z. Qiao, Angew. Chem. Int. Ed. 54, 52–65 (2015)
Z.W. Seh, J. Kibsgaard, C.F. Dickens, I. Chorkendorff, J.K. Nørskov, T.F. Jaramillo, Science 355, 6321 (2017)
A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan, X. Zhang, G. Yang, H. Fu, Nano Energy 44, 353–363 (2018)
A.K. Sun, Y.L. Shen, Z.Z. Wu, D.Z. Wang, Int. J. Hydrogen Energy 42, 14566–145719 (2017)
W.C. Sheng, M. Myint, J.G. Chen, Y.S. Yan, Energy Environ. Sci. 6, 1509 (2013)
W. Zhang, Z. Yu, H.L. Jiang, S.H. Yu, J. Am. Chem. Soc. 136, 14385–14388 (2014)
P. Pachfule, D. Shinde, M. Majumder, Q. Xu, Nat. Chem. 8, 718–724 (2016)
J. Tang, R.R. Salunkhe, J. Liu, N.L. Torad, M. Imura, S. Furukawa, Y. Yamauchi, J. Am. Chem. Soc. 137(4), 1572–1580 (2015)
F.L. Wang, X.D. Yang, B.X. Dong, X. Yu, H.G. Xue, L.G. Feng, Electrochem. Commun. 92, 33–38 (2018)
X.S. Guo, Z.J. Feng, Z.Z. Lv, Y.F. Bu, Y.L. Liu, L.Q. Zhao, C.C. Hao, G.C. Li, Q.Q. Lei, ChemElectroChem 4, 2052–2058 (2017)
P.W. Menezes, A. Indra, C. Das, C. Walter, C. Göbel, V. Gutkin, D. Schmeiβer, M. Driess, ACS Catal. 7, 103–109 (2017)
J. Ren, Z. Hu, C. Chen, Y.P. Liu, Z.Y. Yuan, J. Energy Chem. 26, 1196–1202 (2017)
Y.P. Zhu, Y.P. Liu, T.Z. Ren, Z.Y. Yuan, Adv. Funct. Mater. 25, 7337–7347 (2015)
J.T. Zhang, X. Liang, X.P. Wang, Z.B. Zhuang, Mater. Lett. 202, 146–149 (2017)
X. Liang, D.Z. Zhang, Z.Z. Wu, D.Z. Wang, Appl. Catal. A 524, 134–138 (2016)
Z.H. Pu, I.S. Amiinu, M. Wang, Y.S. Yang, S.C. Mu, Nanoscale 8, 8500–8504 (2016)
Y.F. Zhang, J. Yang, Q.C. Dong, H.B. Geng, Y. Zheng, Y.L. Liu, W.J. Wang, C.C. Li, X.C. Dong, ACS. Appl. Mater Interfaces 10, 26258–262639 (2018)
J. Yang, F.J. Zhang, X. Wang, D.S. He, Angew. Chem. Int. Ed. 128, 13046–13050 (2016)
P. Liu, J.A. Rodriguez, J. Am. Chem. Soc. 127, 14871–14878 (2005)
R. Song, B. Luo, J.F. Geng, D.X. Song, D.W. Jing, Ind. Eng. Chem. Res. 57, 7846–7854 (2018)
X.Y. Li, X. Qian, Y.L. Xu, H. Wu, Y.Y. Dan, L.Z. Chen, Q. Yu, Electrochim. Acta 321, 134646 (2019)
K.L. Sun, K.H. Wang, T.P. Yu, X. Liu, G.X. Wang, L.H. Jiang, Y.Y. Bu, G.W. Xie, Int. J. Hydrogen Energy 44, 1328–1335 (2019)
X.Z. Fang, L. Jiao, R. Zhang, H.L. Jiang, A.C.S. Appl, Mater. Interfaces 9, 23852–23858 (2017)
R. Mo, S. Wang, H.X. Li, J. Li, S. Yang, J.X. Zhong, Electrochim. Acta 290, 649–656 (2018)
Y.M. Du, Y. Han, X.D. Huai, Y.R. Liu, C.Y. Wu, Y. Yang, Int. J. Hydrogen Energy 43, 22226–22234 (2018)
Y.M. Tian, J. Yu, H.S. Zhang, C. Wang, M.L. Zhang, Z.W. Lin, J. Wang, Electrochim. Acta 300, 217–224 (2019)
X.P. Liu, S.F. Deng, D.D. Xiao, M.X. Gong, J.N. Liang, T.H. Zhao, T. Shen, D.L. Wang, A.C.S. Appl, Mater. Interfaces 11, 42233–42242 (2019)
X.P. Yin, D. Lu, J.W. Wang, X.L. Lu, ChemCatChem 11, 5407–5411 (2019)
K.W. Wang, J.S. Ten, Z.J. Lu, S. Chen, X.L. She, H.W. Zhang, D.J. Yang, Int. J. Hydrogen Energy 43, 13939–13945 (2018)
Z. Wang, P. Ju, Y. Zhang, F.H. Jiang, H.B. Ding, C.J. Sun, Electrochim. Acta 187, 424 (2020)
G.S. Song, S. Sun, Z.C. Wang, C.Z. Luo, C.X. Pan, Acta Metall. Sin. (Engl. Lett.) 30, 1008–1016 (2017)
J. Zhao, Z.J. Yin, B.W. Wang, Z.H. Li, Y. Xu, X.B. Ma, J. Energy Chem. 47, 248–255 (2020)
W.P. Xiao, L. Zhang, D. Bukhvalov, Z.P. Chen, Z.Y. Zou, L. Shang, X.F. Yang, D.Q. Yan, F.Y. Han, T.R. Zhang, Nano Energy 70, 104445 (2020)
H.T. Du, L. Xia, S.Y. Zhu, F. Qu, F.L. Qu, Chem. Commun. 23, 2854–2857 (2018)
E.H. Eladgham, D.D. Rodene, R. Sarkar, I.U. Arachchige, R.B. Gubta, A.C.S. Appl, Nano Mater. 3, 8199–8207 (2020)
Y.D. Lu, Y.Y. Deng, S.L. Lu, Y.Y. Liu, J.P. Lang, X.Q. Cao, H.W. Gu, Nanoscale 11, 21259–21265 (2019)
J. Yu, Q.Q. Li, Y. Li, C.Y. Xu, L. Zhen, V.P. Dravid, J.S. Wu, Adv. Funct. Mater. 26, 7644–7651 (2016)
X.L. Yang, A.Y. Lu, Y.H. Zhu, M.N. Hedhili, S.X. Min, K.W. Huang, Y. Han, L.J. Li, Nano Energy 15, 634–641 (2015)
C. Lin, Z.F. Gao, J.H. Yang, B. Liu, J. Jin, J. Mater. Chem. A 6, 6387–6392 (2018)
Y.M. Du, M.J. Zhang, Z.C. Wang, Y.R. Liu, Y.J. Liu, Y.L. Geng, L. Wang, J. Mater. Chem. A 7, 8602–8608 (2019)
P.P. Zhou, Y.Q. Zhang, B. Ye, S. Qin, R.R. Zhang, T.Y. Chen, H.J. Xu, L. Zheng, Q.H. Yang, ChemCatChem 11, 6086–6091 (2019)
K.L. Yan, J.F. Qin, Z.Z. Lin, B. Dong, J.Q. Chi, W.K. Gao, J.H. Lin, Y.M. Chai, C.G. Liu, Chem. Eng. J. 334, 922–931 (2018)
T.Y. Chen, R.R. Zhang, B. Ye, Q.H. Yang, H.J. Xu, L. Zheng, L.G. Wang, Nanotechnology 31, 125402 (2020)
C. Wu, Y.J. Yang, D. Dong, Y.H. Zhang, J.H. Li, Small 13, 1602873 (2017)
W. Zhang, J. Liang, L. Wang, Z.J. Feng, T.T. Yu, Z.H. Zhang, T.T. Yu, Z.H. Zhang, Y.L. Shao, C.C. Hao, G.C. Li, Int. J. Hydrogen Energy 43, 2034–2042 (2018)
Acknowledgements
This work was supported by Anhui Provincial Natural Science Foundation (Grant No. 1808085ME143) and the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Grant No. 2021-KF-20, Wuhan University of Technology).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competitive economic interests or personal relationships, which could affect the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, TY., Zhang, YQ., Fu, YY. et al. Construction of Ni–Mo–P heterostructures with efficient hydrogen evolution performance under acidic condition. J Mater Sci: Mater Electron 32, 14966–14975 (2021). https://doi.org/10.1007/s10854-021-06048-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06048-5