
Abstract
In the present paper, we have investigated the effect of plasmonic gold nanoparticles (Au NPs) decoration on the photocatalytic efficiency of molybdenum disulfide (MoS2) nanosheets. The Au NPs are grown on the surfaces of chemically exfoliated MoS2 nanosheets by chemical reduction method with four different concentrations. The resulting Au-MoS2 nanostructures (NSs) are then characterized by X-ray diffractometer, Raman spectrometer, absorption spectrophotometer, field emission scanning electron microscopy, energy dispersive X-ray, and transmission electron microscopy (TEM). Sizes of the exfoliated MoS2 nanosheets are ~ 700 nm. In addition, the sizes of Au nanoparticles increase from 8.02 ± 2.03 nm to 9.81 ± 3.18 nm with the increase in concentrations of Au ions, as revealed by TEM imaging. Exfoliated MoS2 and Au-MoS2 NSs are used to study the photocatalytic degradation of organic dyes, methyl red (MR) and methylene blue (MB). Under UV–Visible light irradiation, pristine MoS2 shows photodegradation efficiencies in the range of 30.0% to 46.9% for MR, and 23.3% to 44.0% for MB, with varying exposure times of 30 to 120 min. However, Au-MoS2 NSs with the sets having maximum Au NPs concentrations, show enhanced degradation efficiencies from 70.2 to 96.7% for MR, and from 65.2 to 94.3% for MB. The degradation rate constants vary from − 0.5660 to − 1.5551 min−1 for MR dye, and vary from − 0.3587 to − 1.2614 min−1 for MB dye. The multi-fold enhancements of degradation efficiencies for both the dyes with Au-MoS2 NSs, can be attributed to the presence of Au NPs acting as charge trapping sites in the NSs. We believe this type of study could provide a way to battle the ill-effects of environmental degradation that pose a major threat to humans as well as biodiversity. This study can be further extended to other semiconducting materials in conjugation with two dimensional materials for photocatalytic treatment of polluted water.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Synthetic dyes are being excessively used in textile, leather and other industries. These dyes contain organic contaminants that are toxic in nature, and are not completely degraded before getting released as effluents in wastewater, thereby increasing toxicity of water bodies as well as leading to harmful effects on humans and biodiversity. Photocatalysis is an advanced oxidative process (AOP), which leads to quick formation of hydroxyl radicals that has the capability of oxidizing wide range of pollutants present in water without any preference of selectivity [1, 2]. Even if complete mineralization of these contaminants or organic pollutants is not possible, this AOP at least converts them into intermediate compounds that are further biodegradable. Thus, to tackle the menace of environmental degradation, it is often necessary to develop photocatalysts which are non-toxic, potentially stable and cost-effective for large scale degradation of organic contaminants present in water [3]. Traditionally, semiconductor mediated photocatalysts such as TiO2, ZnO, ZnS, SnO2, CdS, CuS, etc. [4,5,6,7,8,9,10] have been studied widely, for complete mineralization of organic pollutants. These semiconductors show high responsivity towards photocatalytic process [11,12,13,14,15,16,17,18]. However, fast recombination of electron–hole pairs in these semiconductors, and the limited responsivity towards UV excitation, restrict their applications as good photocatalysts [2, 19,20,21]. In a typical photocatalytic process, excitation by photons creates charge separation causing the electrons to overcome the band gap of the material. An ideal photocatalyst requires increased surface area for interfacial charge transfer, tailored visible light absorption, tunable band gap, enhanced defect sites for trapping of charge carriers, etc., to enhance the efficiency of the photocatalytic process [1, 12]. Thus, it has become the need of the hour to develop an alternative candidate for a photocatalyst, which can overcome the limitations of pristine semiconductor photocatalysts and can also show improved degradation capabilities.
In recent years, molybdenum disulphide (MoS2), a well-known layered transition metal dichalcogenide (TMDC), has gained significant attention in the field of photocatalysis. MoS2 exhibits high chemical reactivity, varied optical properties, enhanced surface area, charge carrier mobility and strong absorption of visible light [10, 22]. Its flexible band gap from 1.3 eV in bulk to 1.9 eV in monolayer, exhibits an indirect to direct transition, making itself suitable for light absorption in a wide spectrum [23]. Unlike multi-layered materials, presence of tightly bound excitons in monolayer MoS2 produces favourable light absorption properties within direct band gap, making it a visible light assisted photocatalyst [24,25,26]. However, monolayer MoS2 has very low optical cross-section, low number of active sites and fast electron–hole recombination rate, which hinder extensive light-matter interactions leading to weak absorption. In order to overcome fast recombination rate, MoS2 is often incorporated with other semiconducting or noble metal nanoparticles. The incorporated material provides an additional path to the photogenerated electrons and holes, which hinders the recombination rate and thereby enhances the photocatalytic efficiency [27,28,29]. Similar to noble metals like Ag, Pd, Pt, etc., Au NPs also enjoy the advantageous non-toxicity and excellent stability. Au NPs are formed on defect sites of MoS2 and localized by non-covalent bonds. The Au NPs act as a p-type dopant in the MoS2 layer. This leads to strong chemical bonding along the interplanar directions acting as a spacer between interlayers of MoS2, thereby prohibiting restacking of layers [3, 19, 30, 31]. Therefore, deposition of Au NPs on MoS2 nanosheets serves as a sink for electrons, hindering the electron–hole recombination and facilitating interfacial transfer of electrons. This enhancement of charge transportation between Au NPs and MoS2 increases the stability of resulting nano-composites.
There are numerous studies on the growth of metal nanoparticles on MoS2 and the application in sensing and photovoltaic devices. But there are very few studies on the photocatalytic application of metal, especially Au NPs decorated MoS2 nanostructures [4, 19, 32, 33]. In the present paper, we investigate the effect of plasmonic Au NPs decoration on the photocatalytic efficiency of MoS2 nanosheets. The Au NPs are grown on the surface of chemically exfoliated MoS2 nanosheets by solvent-surfactant assisted liquid exfoliation chemical reduction method. N-Methyl 2-pyrrolidone (NMP) is used as a reducing agent for rapid reduction of metal ion (Au3+) to zero-valent gold metal (Au0) in a very short time period. Sodium dodecyl benzene sulfonate (SDBS) is used as surfactant to provide stable dispersions [34]. This solvent-surfactant assisted sonication method helps in avoiding restacking of exfoliated sheets that remain stable over many weeks. These nanostructures are then characterized by XRD, Raman spectrometer, UV–Vis absorption spectrophotometer, field emission scanning electron microscopy, energy dispersive X-ray and transmission electron microscopy, and are used for studying the photocatalytic degradation of organic dyes — methyl red (MR) and methylene blue (MB).
2 Experimental
2.1 Materials
Molybdenum disulfide (MoS2) powder, n-methyl 2- pyrrolidone (NMP), isopropyl alcohol (IPA), hydrogen tetrachloroaurate trihydrate (HAuCl4.3H2O), sodium dodecyl benzene sulfonate (SDBS), methyl red (MR) and methylene blue (MB) dyes were obtained from Sigma Aldrich. All the chemicals were used without further purification. Double distilled water was used as solvent at various stages.
2.2 Chemical exfoliation of MoS2 nanosheets
Synthesis of MoS2 nanosheets have been done via liquid-phase exfoliation method. For the synthesis of MoS2 nanosheets, 20 mL of NMP, IPA and distilled water (volume ratio 3:1:1) mixed solvent was taken and 0.05 g of bulk MoS2 powder was dispersed in it. This mixture was ultrasonicated at room temperature continuously for 24 h. After 24 h of sonication, solution colour changes from dark greyish black to greenish. The resultant green dispersion obtained was kept for 30 min so that the un-exfoliated MoS2 settles down. Thereafter, 18 mL of clear supernatant was collected, and five different sets each of 2 mL of supernatant were taken out and 10 mg of SDBS was added in each set. The solution was then allowed to settle down for another 30 min.
2.3 Synthesis of Au-MoS2 nanostructures
For synthesis of Au-MoS2 nanostructures, a stock solution of 10 mM Au salt (HAuCl4.3H2O) was prepared. Four sets each of 2 mL dispersion of exfoliated MoS2 nanosheets were taken for synthesis of Au-MoS2 nanostructures. In these four sets, different quantities of Au salt solution (2µL, 4µL, 6µL and 8µL) were added and the mixtures were kept for aging for 48 h; these samples were named SET-I, SET-II, SET-III and SET-IV, respectively. Thus the Au-MoS2 nanostructures with varying concentrations of Au NPs were synthesized.
2.4 Characterization techniques
The crystalline structure of as-prepared samples were determined by a powder X-ray diffractometer (XRD) with CuKα radiation (λ = 0.154 nm) in the 2θ range of 20° to 70°. The absorption spectra of MoS2 and Au-MoS2 nanostructures were recorded using absorption spectrophotometer. The absorption spectrophotometer (Labtronics, LT-2700, India) was used for the photocatalytic study as well. The vibrational modes of MoS2, and the effect of Au NPs on these modes, were investigated using a confocal Raman spectrometer having an excitation source of 532 nm laser. The morphologies of MoS2 and Au-MoS2 were analysed by transmission electron microscope JEOL 2100 Plus operated at 200 kV, and field emission scanning electron microscopy (JSM6100 JEOL). The elemental analysis of the pristine MoS2 and Au-MoS2 was done using energy dispersive X-ray (EDX) measurement, carried out on XMAX 80 T (Oxford Instrument Company).
2.5 Photocatalytic test for dye degradation
Methyl red (MR) and methylene blue (MB) are chemically stable and poorly biodegradable dye contaminants in wastewater. In this work, we have used these dyes (MR and MB) as model reactions to evaluate the photocatalytic activity of as-prepared photocatalysts — MoS2 and Au-MoS2 nanostructures. The dye concentration used in this process was 50 mL of 100 ppm aqueous solution of MR and MB. The dye-solutions were treated with different sets of photocatalysts in dark, as well as under UV–visible light of Xenon lamp (1200 W). A fixed quantity of 1 mL of MoS2 dispersion and different sets of 1 mL Au-MoS2 NSs were used as photocatalysts. In presence of photocatalysts, the dye solutions were exposed for 30, 60, 90, and 120 min by using Xenon lamp. After exposure, the solutions were centrifuged for 10 min at 4000 rpm to remove the residuals of the photocatalysts. Thus obtained dye solutions were used to evaluate the photocatalytic properties of the nanostructures.
3 Results and discussion
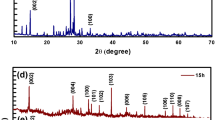
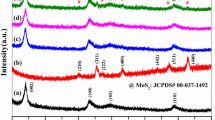
X-ray diffraction was used to characterize the crystalline structure and to count the microstructures of material in large quantities. Figure 1 shows the X-ray diffraction patterns of Au, MoS2 and Au-MoS2 NSs.

XRD patterns of the Au, MoS2 and Au-MoS2 nanostructures (SET-I to SET-IV). (Asterisk represents Au and black filled square indicates MoS2 XRD patterns)
In these XRD patterns, diffraction peaks at 29.19°, 32.88°, 33.68°, 36.09°, 39.75°, 44.34°, 49.99°, 56.19°, 58.50°, 60.61° and 62.95° are observed which are attributed to the lattice planes (004), (100), (101), (102), (103), (006), (105), (106), (110), (008) and (107) of MoS2 respectively (JCPDS No. 37–1492) [35]. After Au NPs decoration, the Au-MoS2 NSs showed extra diffraction peaks at 37.34°, 44.17° and 64.40° corresponding to the (111), (200) and (220) planes, respectively of Au phase (JCPDS No. 04–0784) [36] indicating that FCC Au NPs had been successfully decorated on MoS2 surface. A slight shift in the peak positions of synthesized MoS2 and Au-MoS2 nanostructures, as compared to that of standard XRD peaks was observed, which is due to variation in conditions of both the cases. As observed in Fig. 1, the intensity of Au peak enhances as we increase the concentration of Au, while the intensity of peaks of MoS2 decreases accordingly in SET-I, SET-II, SET-III and SET-IV (with increasing concentration of Au—2µL, 4µL, 6µL and 8 µL, respectively). Lowering in diffraction peak intensity is due to decrease in the exposure area of MoS2 nanosheets in presence of Au NPs. The crystallite size of MoS2 is found to be 44.18 nm in case of pristine, while in the SET-I, SET-II, SET-III, and SET-IV, the values come out to be 39.01 nm, 37.34 nm, 35.85 nm and 34.88 nm, respectively, as calculated by using Debye Scherrer’s formula [37],
where D is the mean size of the crystallites, k is a dimensionless shape factor (~ 0.9), \(\lambda \) is the X-ray wavelength (λ = 1.5418 Å), \(\beta \) is the line broadening at full width at half maximum (FWHM), \(\theta \) is the position of the diffraction peak. As the dimension decreases, peaks broaden, which shows that the NSs lose their crystallinity with the resultant increase in amorphous to crystalline fraction ratio. Thus, size-dependent broadening and intensity of patterns can be attributed to reduced crystallite size, in-built strain in the MoS2 lattice due to edge effects, and loss of crystalline fraction in small-sized NSs. The lattice strain in MoS2 was calculated using peak broadening analysis. The lattice stain ε is given by following equation [37]
Williamson-Hall (W–H) equation represents size-induced and strain-induced broadening in the XRD peaks, which is given by
The Williamson-Hall plots of pristine MoS2 and Au-MoS2 are shown in Fig. 2.

Williamson-Hall plots for pristine MoS2 and Au-MoS2 nanostructure. The linear variation of \(\beta \,cos\,\theta \) with respect to \(sin\theta \) is depicted by fitting straight lines
The W–H analysis shows that the lattice strain in MoS2 is of the order of 0.0018, while for Au-MoS2 it is 0.0015.
The optical absorptions of pristine MoS2 nanosheets and Au-MoS2 nanostructures have been explored using UV–Vis absorption spectrophotometer. The absorption spectra of MoS2 and Au-MoS2 are shown in Fig. 3. The absorption spectrum of MoS2 nanosheet shows two small humps in the visible range, one at 670 nm and other at 620 nm, which are known as A and B excitonic peaks, respectively. These peaks arise due to spin–orbit interactions causing splitting of valence band energy levels, and the excitonic transitions between split valence bands and minima of conduction band at the K-point of the Brillouin zone [38]. Interlayer coupling also plays an important role in valence band splitting. When a small aliquot of gold precursor is added into chemically exfoliated MoS2, a new absorption peak corresponding to the Au plasmon band emerges at around 530 nm, suggesting consumption of Au3+ ions and formation of gold nanoparticles. The extent of Au-ion reduction by the spontaneous redox reaction can be estimated by monitoring the absorption peak of Au3+ and quantifying the loading level of Au on the surface of MoS2. As we increase the Au concentration, the surface plasmon resonance (SPR) peak of Au experiences both red shift and increase in intensity (as shown in Fig. 3). The shift infers strong plasmon-excitonic coupling between Au and MoS2. The surface plasmon resonance strongly depends on shape, size and separation of the nanoparticles along with the surrounding environment. In Fig. 3, it is observed that when concentration of Au is low, the SPR peak is not so prominent, which is due to smaller size and low concentration of Au NPs.

Absorption spectra of MoS2 and Au-MoS2 nanostructures with different concentrations of Au
Raman spectroscopy was used to investigate the crystallinity and layer thickness of two dimensional (2D) MoS2 in terms of the position and frequency difference of two characteristic vibrational modes, E12g and A1g. It is also used for finding the effects of lattice strain, doping levels, and the van der Waals interaction at the interface of 2D crystals [39]. The E12g mode is attributed to the in-plane vibration of Mo and S atoms, and this mode is sensitive to the built-in strain of 2D MoS2. The A1g mode is related to the out-of-plane vibration of S atoms which is a reflection of interlayer van der Waals interaction.
The Raman spectra of MoS2 shown in Fig. 4 exhibit two vibrational modes centred at 386.03 cm−1 and 411.22 cm−1, respectively. In case of Au-MoS2 NSs, red shifts in E12g and A1g modes are observed. The SET-IV, with maximum loading of Au NPs, exhibits red shifts of E12g and A1g modes; these peaks are observed at 384.64 cm−1 (shift ~ 1.39 cm−1) and 410.14 cm−1 (shift ~ 1.08 cm−1), respectively. The shifting is attributed to the effect of lattice strain due to curvature of the MoS2 shell. The frequency difference of E12g and A1g peaks for MoS2 comes out to be about 25.19 cm−1 which renders exfoliation of few-layers MoS2 sheets [23]. Similar frequency difference was observed for Au-MoS2 sheets, i.e., 25.50 cm−1 for SET-IV. Besides variation in Raman frequency, we have also observed a significant enhancement of Raman peak intensity in the Au-MoS2 nanostructures. This can be attributed to the effect of localized surface plasmon resonance (LSPR) of Au nanoparticle, typically called surface enhanced Raman scattering (SERS) [40]. Interaction of incident light with Au NPs excites localized surface plasmons. When there is resonance between the frequency of plasmon oscillation and irradiation, a strong electromagnetic field is formed on the surface. This electromagnetic field leads to a significant increase in the intensity of Raman mode as shown in Fig. 4. The intensity ratio of A1g/ E12g, for exfoliated MoS2 is found to be 1.04. However with the incorporation of Au NPs, the peak intensity ratio increases from 1.23 (SET-I) to 1.30 (SET-IV). Previous study had shown that the increase in A1g/E12g peak intensity ratio is due to the p-type doping caused in MoS2 by Au NPs decoration [41].

Raman spectra of MoS2 and Au-MoS2 nanostructures
The shapes and sizes of MoS2 and Au-MoS2 nanostructures were investigated using transmission electron microscopy (TEM). The TEM images of MoS2 and Au-MoS2 nanostructures along with their selective area electron diffraction (SAED) patterns are shown in Fig. 5.

TEM images of a MoS2 and c Au-MoS2 nanostructures (SET-IV) along with their selective area electron diffraction patterns in b and d, respectively; the scale bar in a is equivalent to 500 nm and in c it is equivalent to 200 nm
In Fig. 5a sheet-like structure is observed, which corresponds to pristine MoS2. Sizes of these nanosheets are ~ 700 nm. In MoS2, lattice spacing is of the order of 0.27 nm, which corresponds to (100) plane. The SAED pattern of MoS2 nanosheet is shown in Fig. 5b. It consists of many bright spots, which correspond to hexagonal lattice structure of MoS2. In the present case, MoS2 is in few-layered structure and each layer forms a hexagonal diffraction pattern; hence many spots are observed. In case of Au-MoS2, Au nanoparticles are found to be of spherical shape of size 9.81 ± 3.18 nm, formed on the edges of nanosheets along with some agglomerations of these particles (Fig. 5c). The lattice plane spacing in the Au nanoparticles corresponding to the FCC lattice of Au is of the order of 0.22 nm, which corresponds to (111) plane of gold lattice. The SAED pattern of Au-MoS2 is shown in Fig. 5d. It shows diffraction patterns corresponding to Au and MoS2. In addition, SET-I is explored under HRTEM. Figure S2 (in Supporting Information) depicts histogram of Au-MoS2 nanostructures (SET-I) revealing that the synthesized Au nanostructures are of spherical shape with particle size of 8.02 ± 2.03 nm. With the increase in concentration of Au ions, there is an increase in size (~ 2 nm) with particle size 9.81 ± 3.18 nm, as depicted from the histogram (Fig. S4 in Supporting Information).
Morphology of MoS2 and Au-MoS2 nanostructures were further investigated using field emission scanning electron microscope (FESEM). The FESEM images of pristine MoS2 and Au-MoS2 (SET-IV) are shown in Fig. 6. The sheet-like structure of MoS2 is depicted in Fig. 6a. From FESEM image of MoS2, it is observed that the nanosheets consist of few layers and these nanosheets are of varying sizes. Figure 6b shows FESEM image of Au-MoS2 nanostructures. In this image, it is observed that there are additional particles of Au of different sizes, on the edge of MoS2 nanosheets. In case of FESEM, there is increase in size (~ 40 nm) as compared to TEM results, which might be due to the agglomeration of Au nanoparticles and forming clusters.

The FESEM images of a pristine MoS2 nanosheets and b Au-MoS2 nanostructures
The elemental analysis of pristine MoS2 and Au-MoS2 nanostructures was carried out by elemental mapping of the samples. The elemental mappings of these samples are shown in Fig. 7. Figure 7a–c corresponds to pristine MoS2. In Fig. 7a two distinct coloured spots are observed. The green spots correspond to Mo elements present in the sample, and red spots correspond to S elements. These Mo and S elements are shown in Fig. 7b and c, respectively, as well. Figure 7d–g represent the elements present in Au-MoS2 nanostructures. Here, three different coloured spots are observed, which correspond to three distinct elements, Au, Mo and S, respectively.

The elemental mapping of a-c MoS2 and d-g Au-MoS2 nanostructures
The EDX spectra of pristine MoS2 and Au-MoS2 are shown in Fig. 8. The spectra consist of peaks corresponding to the elements present in MoS2 and Au-MoS2. In EDX spectra, additional peak around 8 keV is observed, which is due to copper grid.

Energy dispersive X-ray spectra of a MoS2 and b Au-MoS2 nanostructure (SET-IV)
The as-synthesized MoS2 and Au-MoS2 NSs were used for photocatalytic degradation of dye molecules. A blank test in the absence of catalysts was done and negligible self-degradation of dyes was observed.
The characteristic absorption peak of MR appears at 523 nm. Figure 9a–e show the absorption spectra of MR dye molecules under UV–Visible light irradiation, in presence of MoS2; SET-I, SET-II, SET-III and SET-IV are for different exposure times (30 -120 min). A systematic decrease in the absorbance of the dye molecules is observed with increasing exposure time in presence of MoS2 (Fig. 9a). This is due to the degradation of MR dye molecules. The degradation is attributed to electron–hole pair generation in MoS2 nanosheets. These electron–hole pairs interact with oxygen and water molecules, and thereby create reactive oxygen species, which further interact with the dye molecules and decompose them. In case of pristine MoS2, photodegradation efficiency is limited due to direct band gap of MoS2. The direct bandgap causes fast recombination of photogenerated electron–hole pairs in MoS2. The degradation efficiency of MoS2 can be enhanced by incorporating Au nanoparticles in the MoS2 as presented in Fig. 9b-e. In Fig. 9b–e, with incorporation of Au nanoparticles, the characteristic peak of MR shows downshift (hypochromic shift) as concentration of Au increases in Au-MoS2 NSs. SET-IV exhibits maximum decrease in absorbance of the dye molecules. The concentration of dye was monitored with the aid of absorption spectroscopy. According to Beer-Lambert’s law, absorbance is proportional to concentration of dye molecules, so its percentage degradation efficiency can be calculated by following equation [30]

Absorption spectra of MR after exposure of UV–Vis light for different time in presence of a MoS2 nanostructures, b SET-I, c SET-II, d SET-III and e SET-IV. (SET I-SET IV, increasing concentration of Au from 2 µL to 8 µL with the difference of 2µL)
where Co, C are concentrations and Ao, A are absorbances of dye at the initial time and after final time t, respectively. For quantitative understanding of the effect of photocatalysts on reaction kinetics of the dyes, we have utilized the pseudo-first order reaction mechanism model. The rate constant k of the degradation was investigated using
where Co is the initial concentration, C is the concentration at t exposure time, k is the pseudo-first-order rate constant. The rate constants are obtained from the regression line between ln(C/Co) and time t. Figure 10a represents the percentage degradation of MR with MoS2, with SET-I, SET-II, SET-III and SET-IV as photocatalysts. It is found that degradation efficiency of MoS2 enhances from 30.0 to 46.9%, while varying the exposure time from 30 to 120 min. For SET-I, the degradation efficiency enhances from 37 to 68%, for SET-II it enhances from 45.0 to 80.2%, for SET-III from 55.0 to 89.3% and for SET-IV the degradation efficiency enhances from 70.2 to 96.7%, after irradiating with UV–Visible light and varying the exposure time from 30 to 120 min. Thus, we observe that with increase in Au concentration from SET-I to SET-IV and varying the exposure time from 30 to 120 min, the degradation efficiency increases. For longer exposure to UV–Visible light irradiation (120 min), more electron–hole pairs are generated, which causes enhanced degradation of dye molecules.

a Percentage degradation of MR with MoS2, SET-I, SET-II, SET-III and SET-IV, b kinetic graph of MR with photocatalysts MoS2 and different sets of Au-MoS2
The reaction kinetics corresponding to photocatalytic degradation of MR are plotted in Fig. 10b. The rate constant values calculated from the regression lines of MR for MoS2, SET-I, SET-II, SET-III and SET-IV are − 0.5660 min−1, − 0.7996 min−1, − 1.0693 min−1, − 1.2832 min−1 and − 1.5551 min−1, respectively. This enhancement in the magnitude of the rate constant of the photocatalysts for MR signifies major degradation of dye by SET-IV, as compared to other photocatalysts. The percentage degradation of dye by MoS2, SET-I, SET-II, SET-III and SET-IV along with rate constant values for 120 min of irradiation time are compiled in Table 1.
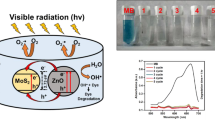
A pictorial image of MR dye under different photocatalytic treatment is presented in Fig. 11. From image, it is clearly observed that increasing the Au NPs concentration and the exposure time of UV–Visible light irradiation, cause the colour of the dye to fade out, which reflects degradation of dye molecules.

Pictorial representation of MR degradation in 0, 30, 60, 90, and 120 min with MoS2, SET-I, SET-II, SET-III and SET-IV (SET-I to SET-IV increasing conc. of Au in MoS2 by the factor 2µL)
Similar to MR dye molecules, we have also investigated the photodegradation of MB dye using MoS2 and different sets of Au-MoS2 NSs as catalysts, under UV–Visible light irradiation. The characteristic peak of MB is monitored at 663 nm by using a UV–Visible spectrophotometer. In Fig. 12a, with MoS2 as photocatalyst, a downshift in the absorbance curve is observed with increase in irradiation time. The change in absorbance is moderate, which indicates less efficiency towards dye decolorization due to confined band gap of MoS2. Whereas, when different sets of Au-MoS2 NSs are used as photocatalysts, as shown in Fig. 12b–e, a noticeable decrease in absorbance with time is observed. From the absorption data it is clear that coupling of Au with MoS2, especially for SET-IV among all four sets, provides better degradation efficiencies as compared to MoS2 and other sets of Au-MoS2.

Absorption spectra of MB after exposure of UV–Vis light for different time in presence of a MoS2 nanostructures, b SET-I, c SET-II, d SET-III and e SET-IV
To examine the percentage degradation of dye by catalysts, Eq. (4) is applied and results are shown in Fig. 13a. Figure 13a illustrates increase in percentage degradation efficiency with different catalysts. For MoS2, it increases from 23.3 to 44.0%; for SET-I from 32.0 to 65.2%; for SET-II from 39.2 to 78.2%; for SET-III from 52.0 to 88.0%; and for SET-IV the efficiency increases from 65.2 to 94.3% with exposure times varying from 30 to 120 min. These results of percentage degradation confirm that SET-IV exhibits much better degradation efficiency as compared to other catalysts. The reaction kinetics corresponding to photocatalytic degradation of MB are plotted in Fig. 13b. The rate constant values obtained from the regression lines of MB are − 0.3587 min−1 for MoS2, − 0.5612 min−1 for SET-I, -0.7490 min−1 for SET-II, − 0.9689 min−1 for SET-III, and − 1.2614 min−1 for SET-IV. The increase in the magnitude of rate constants of photocatalysts for MB indicates major elimination of dye by SET-IV. The percentage degradation of dye with different photocatalysts (MoS2, SET-I, SET-II, SET-III and SET-IV) and consequent rate constant values are compiled in Table 2.

a Percentage degradation of MB with MoS2, SET-I, SET-II, SET-III and SET-IV, b Kinetic graph of MB with photocatalysts MoS2 and different sets of Au-MoS2
Figure 14 illustrates the pictorial images of MB degradation with different irradiation times. It is observed that under UV–Visible light irradiation, the dye colour changes. SET-IV exhibits maximum fading of colour, which correlates with lower absorption spectra in Fig. 12e corresponding to maximum degradation of MB in 120 min.

Pictorial representation of MB degradation in 120 min with different photocatalysts (MoS2, SET-I, SET-II, SET-III and SET-IV; SET-I to SET-IV increasing concentration of Au in MoS2 by factor 2µL)
Due to enlarged surface area, two dimensional MoS2 nanosheets enhance the interfacial charge transfer processes between photocatalyst and oxygen molecules dissolved in wastewater. Basal planes of MoS2 are quite stable under photocatalytic activity but the edge sites or defect sites participate actively in the photocatalysis process. Au NPs nucleate at these highly energetic defect sites, such as edges or line defects, and enhance the charge transportation between Au NPs and MoS2. Figure 15a shows the mechanism of charge transfer from MoS2 to Au NPs and Fig. 15b represents the energy level diagram of Au-MoS2 interface. Figure 15b shows that the work function of MoS2 is 5.03 eV whereas for gold it is 4.58 eV. The Fermi level of MoS2 is situated well above the reduction potential (1.50 eV) of Au. The electron–hole pairs are created under UV–Visible light irradiation with photon energy greater than the energy band gap (Eg) of MoS2 (Eq. 6). However, these electron–hole pairs have a tendency of recombining easily. Incorporation of Au NPs in MoS2 nanosheets helps to provide an additional path to the photogenerated electrons in the conduction band (Ec = − 4.10 eV) of MoS2, and move on to the surface of Au NPs (EF = − 4.58 eV). This accelerates efficient splitting of charge carriers, which helps in minimizing possible electron–hole recombination. These Au NPs act as charge trapping centres. The photogenerated holes in the valence band, which survive the process of rapid recombination, diffuse into surface of the catalyst and reacts with water molecules donating electrons to form free hyper-reactive hydroxyl radicals (•OH) (Eq. 7). The surface hydroxyl groups present on the surface of the catalyst act as a trapping sites for holes (h+) from VB and generate hydroxyl radicals. These hydroxyl radicals then react with organic dye molecules or contaminants non-selectively, by adding to their aromatic rings or through double bonds. The resulting compound forms species with carbon centred radicals, which then combine with oxygen molecules to produce compound radicals that are easily transformed into CO2. On the other hand, the photoexcited electrons accumulate on surface of Au NPs and create superoxide radical anions (O2−), which react with oxygen molecules dissolved in wastewater (Eq. 8) [42]. With increase in concentration of Au NPs, the charge trapping centres are also increased, which help to create more holes (h+) in MoS2 and electrons (e−) on the surface of Au. These electrons and holes act with water and oxygen molecules, and create more reactive oxygen species (ROSs). The ROSs help in photo-reduction of organic dyes (Eq. 9) causing degradation of dye molecules. Thus, increasing the concentration of Au NPs helps to enhance the degradation efficiency. On the other hand, holes in the valence band (VB) of MoS2 react with water molecules to form hydroxyl radicals (•OH), which stimulate the degradation of dye molecules (Eq. 10 and Eq. 11) by photo-oxidation process [43].

a Charge transfer mechanism in Au-MoS2 nanostructures and b energy level diagram of the Au-MoS2 interface
The noble metal (Au) is recognized to act as sink for photo-induced charge carriers, promoting interfacial charge transfer processes, and hence enhances the photodegradation efficiency of Au-MoS2 nanostructures [44].
Figure 16a and b represent the molecular structure of MR and MB, respectively. Colour of any dye depends broadly on the chromophoric sites present in that dye. MR and MB differ not only in their molecular structure but also differ in their functional groups attached, and their extent of ionization in any aqueous solution. Both the dyes are basically cationic by nature. Degradation of MR and MB with Au-MoS2 NSs depends upon the number of chromophoric sites present in them, and their destabilization with free radicals generated by photocatalysis mechanism reaction [30]. MR has 1 chromophoric site, while 4 active sites are present in MB. The •OH or O2− radicals generated under irradiation by UV–Visible light destabilize or break the chromophoric sites present in MR and MB [45]. Comparing the molecular structure of MR and MB, MB has 4 methyl groups in addition to these chromophoric sites, which produce more inductive effect and prevent the dye from degradation easily. This leads to lower degradation efficiency for MB as compared to MR.

Molecular structure of a methyl red and b methylene blue
4 Conclusions
In summary, heterostructures of Au-MoS2 nanostructures have been synthesized by the reduction of [AuCl4]− on the surface of MoS2. The Au-MoS2 NSs have been found to improve the efficiency of interfacial charge transfer process in photocatalysis. The photocatalytic studies indicate the decolourization of MR and MB dyes in presence of exfoliated MoS2, and different sets of Au-MoS2 catalysts. It is found that Au-MoS2 NSs improve the efficiencies of photodegradation of organic dyes, MR and MB. The degradation efficiencies increase up to 96.7% and 94.3% for MR and MB, respectively, have been achieved with maximum concentration of Au in Au-MoS2 NSs. The deposited noble metal NPs exhibit interfacial electron transfer, which leads to suppression of recombination of charge carriers. This study provides a detailed insight into the fabrication of UV–Visible light active Au-MoS2 nanostructures with enhanced photocatalytic performance and photo-stability. As a result, Au-MoS2 nanostructures act as favourable candidates for photocatalytic degradation of toxic organic contaminants in wastewater, which is beneficial for environmental remediation concern.
References
K.C. Lalithambika, K. Shanmugapriya, S. Sriram, Photocatalytic activity of MoS2 nanoparticles: an experimental and DFT analysis. Appl. Phys. A 125, 817 (2019)
Z. Li, X. Meng, Z. Zhang, Recent development on MoS2-based photocatalysis: A review. J. Photochem. Photobiol. C Photochem. Rev. 35, 39–55 (2018)
M. Sharma, P.K. Mohapatra, D. Bahadur, Improved photocatalytic degradation of organic dye using Ag3PO4/MoS2 nanocomposite. Front. Mater. Sci. 11, 366–374 (2017)
K. Rani, A.S. Singh, A. Patel, S. Chakraborti, K. Kumar, P. Ghosh, Sharma, Visible light driven photocatalysis of organic dyes using SnO2 decorated MoS2 nanocomposites. Chem. Phys. Lett. 738, 136874 (2020)
S. Kapatel, C.K. Sumesh, Two-step facile preparation of MoS2·ZnO nanocomposite as efficient photocatalyst for methylene blue (Dye) degradation. Electron. Mater. Lett. 15, 119–132 (2019)
M. Mittal, A. Gupta, O.P. Pandey, Role of oxygen vacancies in Ag/Au doped CeO2 nanoparticles for fast photocatalysis. Sol. Energy 165, 206–216 (2018)
E. Benavente, F. Durán, C. Sotomayor-Torres, G. González, Heterostructured layered hybrid ZnO/MoS2 nanosheets with enhanced visible light photocatalytic activity. J. Phys. Chem. Solids 113, 119–124 (2018)
K.M. Reza, A. Kurny, F. Gulshan, Parameters affecting the photocatalytic degradation of dyes using TiO2: A review. Appl. Water Sci. 7, 1569–1578 (2017)
M. Mehrabian, Z. Esteki, Degradation of methylene blue by photocatalysis of cooper assisted ZnS thin film. Optik 130, 1168–1172 (2017)
M. Gerawork, Photodegradation of methyl orange dye by using zinc oxide-copper oxide nanocomposite. Optik 216, 164864 (2020)
K. Zhu, L. Luo, T. Peng, Preparation, Characterization, and Catalytic Performance of MoS2 Photocatalyst. J. Wuhan Univ. Technol. Mater. Sci. Ed. 34: 883–887 (2019)
Z. Fan, F. Meng, M. Zhang, Z. Wu, Z. Sun, A. Li, Solvothermal synthesis of hierarchical TiO2 nanostructures with tunable morphology and enhanced photocatalytic activity. Appl. Surf. Sci. 360, 298–305 (2016)
F.M. Meng, F. Lu, Z.Q. Sun, J.G. Lü, A mechanism for enhanced photocatalytic activity of nano-size silver particle modified titanium dioxide thin films. Sci. China Technol. Sci. 53, 3027–3032 (2010)
Z. Fan, F. Meng, J. Gong, H. Li, Y. Hu, D. Liu, Enhanced photocatalytic activity of hierarchical flower-like CeO2/TiO2 heterostructures. Mater. Lett. 175, 36–39 (2016)
R. Qin, F. Meng, M.W. Khan, B. Yu, H. Li, Z. Fan, J. Gong, Fabrication and enhanced photocatalytic property of TiO2-ZnO composite photocatalyst. Mater. Lett. 240, 84–87 (2019)
F. Yu, M.W. Meng, R. Khan, X. Qin, Liu, Facile synthesis of AgNPs modified TiO2@g-C3N4 hetrojunction composites with enhanced photocatalytic activity under simulated sunlight. Mater. Res. Bull. 121, 110641 (2020)
F. Yu, T. Meng, A. Zhou, Z. Fan, M.W. Zhao, Khan, Construction of CoS/CeO2 hetrostructure nanocages with enhanced photocatalytic performance under visible light. J. Am. Ceram. 103, 6136–6148 (2020)
B. Yu, F. Meng, M.W. Khan, R. Qin, X. Liu, Synthesis of hollow TiO2@ g-C3N4/Co3O4 coreshell microspheres for effective photooxidation degradation of tetracycline and MO. Ceram. Int. 46(9), 13133–13143 (2020)
H. Wang, L. Cui, S. Chen, M. Guo, S. Lu, Y. Xiang, A new perspective on metal particles enhanced MoS2 photocatalysis in hydrogen evolution: Excited electric field by surface plasmon resonance. J. Appl. Phys. 126, 015101 (2019)
A. Bumajdad, M. Madkour, Y. Abdel-Moneam, M. El-Kemary, Nanostructured mesoporous Au/TiO2 for photocatalytic degradation of a textile dye: the effect of size similarity of the deposited Au with that of TiO2 pores. J. Mater. Sci. 49, 1743–1754 (2014)
J. Pan, Z. Jiao, Y. Li, C. Guo, Y. Feng, L. Liu, M. Wang, Wu, Efficient Separation of Electron-Hole Pairs in Graphene Quantum Dots by TiO2 Heterojunctions for Dye Degradation. ACS Sustain. Chem. Eng. 3, 2405–2413 (2015)
S. Mouri, Y. Miyauchi, K. Matsuda, Tunable Photoluminescence of Monolayer MoS2 via Chemical Doping. Nano Lett. 13, 5944–5948 (2013)
T.P. Nguyen, W. Sohn, J.H. Oh, H.W. Jang, S.Y. Kim, Size-Dependent Properties of Two-Dimensional MoS2 and WS2. J. Phys. Chem. C 120, 10078–10085 (2016)
S.V.P. Vattikuti, C. Byon, C.V. Reddy, Improved photocatalytic activity of MoS2 nanosheets decorated with SnO2 nanoparticles. RSC Adv. 5, 86675–86684 (2015)
J. Cheng, L. Han, Y. Wei, Q. Chen, Enhancement of photocatalytic property on ZnS/MoS2 composite under visible light irradiation. MATEC Web Conf. 108, 01008 (2017)
S.V.P. Vattikuti, P.C. Nagajyothi, J. Shim, Use of lactic acid modified MoS2 nanopetals to improve photocatalytic degradation of organic pollutants. Mater. Res. Express 5, 095016 (2018)
X. Yang, H. Yu, X. Guo, Q. Ding, T. Pullerits, R. Wang, G. Zhang, W. Liang, M. Sun, Plasmon-exciton coupling of monolayer MoS2-Ag nanoparticles hybrids for surface catalytic reaction. Mater. Today Energy 7, 72–78 (2017)
A.P. Manuel, A. Kirkey, N. Mahdi, K. Shankar, Plexcitonics- fundamental principles and optoelectronic applications. J. Mater. Chem. C 7, 1821–1853 (2019)
W. Lin, Y. Cao, P. Wang, M. Sun, Unified Treatment for Plasmon-Exciton Co-driven Reduction and Oxidation Reactions. Langmuir 33, 12102–12107 (2017)
R. Singh, P.B. Barman, D. Sharma, Synthesis, structural and optical properties of Ag doped ZnO nanoparticles with enhanced photocatalytic properties by photo degradation of organic dyes. J. Mater. Sci. Mater. Electron. 28, 5705–5717 (2017)
S. Guo, X. Li, J. Zhu, T. Tong, B. Wei, Au NPs@MoS2 Sub-Micrometer Sphere-ZnO Nanorod Hybrid Structures for Efficient Photocatalytic Hydrogen Evolution with Excellent Stability. Small 12, 5692–5701 (2016)
M. Sigiro, Effect of gold dopant on structural and optical properties of molybdenum disulfide single crystals. AIP Conf. Proc. 1855, 030003 (2017)
M. Velický, G.E. Donnelly, W.R. Hendren, S. McFarland, D. Scullion, W.J.I. Debenedetti, G.C. Correa, Y. Han, A.J. Wain, M.A. Hines, D.A. Muller, K.S. Novoselov, H.D. Abruna, R.M. Bowman, E.J.G. Santos, F. Huang, Mechanism of Gold-Assisted Exfoliation of Centimeter-Sized Transition-Metal Dichalcogenide Monolayers. ACS Nano 12, 10463–10472 (2018)
S.H. Lee, D.H. Lee, W.J. Lee, S.O. Kim, Tailored Assembly of Carbon Nanotubes and Graphene. Adv. Funct. Mater. 21, 1338–1354 (2011)
Z. Wu, D. Wang, A. Sun, Surfactant-assisted fabrication of MoS2 nanospheres. J. Mater. Sci. 45, 182–187 (2010)
W.E. Ochanda, Jones, Sub-Micrometer-Sized Metal Tubes from Electrospun Fiber Templates. Langmuir 21, 10791–10796 (2005)
R. Sivakami, S. Dhanuskodi, R. Karvembu, Estimation of lattice strain in nanocrystalline RuO2 by Williamson-Hall and size-strain plot methods. Spectrochim. Acta A 152, 43–50 (2016)
M.D.J. Quinn, N.H. Ho, S.M. Notley, Aqueous dispersions of exfoliated molybdenum disulfide for use in visible-light photocatalysis. ACS Appl. Mater. Interfaces 5, 12751–12756 (2013)
Y. Li, J.D. Cain, E.D. Hanson, A.A. Murthy, S. Hao, F. Shi, Q. Li, C. Wolverton, X. Chen, V.P. Dravid, Au@MoS2 Core-Shell Heterostructures with Strong Light-Matter Interactions. Nano Lett. 16, 7696–7701 (2016)
Z. Li, S. Jiang, Y. Huo, M. Liu, C. Yang, C. Zhang, X. Liu, Y. Sheng, C. Li, B. Man, Controlled-layer and large-area MoS2 films encapsulated Au nanoparticle hybrids for SERS. Opt. Express 24, 26097–26108 (2016)
Y. Shi, J.K. Huang, L. Jin, Y.T. Hsu, S.F. Yu, L.J. Li, H.Y. Yang, Selective Decoration of Au Nanoparticles on Monolayer MoS2 Single Crystals. Sci. Rep. 3, 1839 (2013)
V. Kandavelu, H. Kastien, K.R. Thampi, Photocatalytic degradation of isothiazolin-3-ones in water and emulsion paints containing nanocrystalline TiO2 and ZnO catalysts. Appl. Catal. B Environ. 48, 101–111 (2004)
Marcì, V. Augugliaro, M.J. López-Muñoz, C. Martín, L. Palmisano, V. Rives, M. Schiavello, R.J.D. Tilley, A.M. Venezia, Preparation Characterization and Photocatalytic Activity of Polycrystalline ZnO/TiO2 Systems. 2. Surface, Bulk Characterization, and 4-Nitrophenol Photodegradation in Liquid−Solid Regime. J. Phys. Chem. B 105, 1033–1040 (2001)
J. Shakya, A.S. Patel, F. Singh, T. Mohanty, Composition dependent Fermi level shifting of Au decorated MoS2 nanosheets. Appl. Phys. Lett. 108, 013103 (2016)
A.R. Khataee, M.B. Kasiri, Photocatalytic degradation of organic dyes in the presence of nanostructured titanium dioxide: Influence of the chemical structure of dyes. J. Mol. Catal. A Chem. 328, 8–26 (2010)
Acknowledgements
Authors are thankful to CIL and SAIF Punjab University, Chandigarh for HRTEM, EDX and FESEM measurements, AIRF JNU for Raman measurement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rani, A., Patel, A.S., Chakraborti, A. et al. Enhanced photocatalytic activity of plasmonic Au nanoparticles incorporated MoS2 nanosheets for degradation of organic dyes. J Mater Sci: Mater Electron 32, 6168–6184 (2021). https://doi.org/10.1007/s10854-021-05334-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-05334-6