
Abstract
X-ray diffraction (XRD) techniques are powerful, non-destructive characterization tool with minimal sample preparation. XRD provides the first information about the materials phases, crystalline structure, average crystallite size, micro and macro strain, orientation parameter, texture coefficient, degree of crystallinity, crystal defects etc. XRD analysis provides information about the bulk, polycrystalline thin films, and multilayer structures, which is very important in various scientific and material engineering fields. This review discusses the diffraction related phenomena/principles such as powder X-ray diffraction, and thin-film/grazing incidence X-ray diffraction (GIXRD) comprehensively for thin film samples which are used frequently in various branches of science and technology. The review also covers few case studies on polycrystalline thin-film samples related to phase analysis, preferred orientation parameter (texture coefficient) analysis, stress evaluation in thin films and multilayer, multiphase content identification, bifurcation of multiphase on multilayer samples, depth profiling in thin-film/ multilayer structures, the impact of doping effect on structural properties of thin films etc., comprehensively using GIXRD/XRD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The thin-film characteristics are quite different from the respective bulk counterparts. Materials, having at least one of the dimensions in the nanometer scale (less than 100 nm), are known as nanostructured materials, where properties are significantly different because of geometrical restrictions, causing novel physical and chemical properties. Thin films (either it is polycrystalline, or single crystal (epitaxial) or amorphous form) of a thickness (< 1 µm), possess higher surface energy due to its large surface to volume ratio; thus responsible for different physical, material and chemical properties than their bulk counterpart [1, 2]. This scope of tailoring materials’ to create the desired physical properties led to large research activities in many areas such as microelectronics, optoelectronics, and sensors [3]. In these devices, precise growth of multiple layers of films (in either amorphous, polycrystalline or epitaxial form) of polymers, metals, semiconductors, and insulators is essential to get the desired outcome. These films are deposited using different techniques such as molecular beam epitaxy (MBE), chemical vapor deposition (CVD), pulsed laser deposition (PLD), sputtering, thermal evaporation, e-beam evaporation, electroplating, and spin coating techniques [1, 4]. In all these growth techniques, different characterization techniques involving X-ray radiations (based on scattering, emission, and absorption) are employed to characterize thin films. It is a rapid and non-destructive technique, which requires minimal sample preparation. It provides a wealth of information about the crystalline structure of the sample. The chemical composition of materials can be determined by using X-ray emission spectroscopy. The X-ray’s absorption property is used as an analytical technique based on variation in contrast, like tomography and imaging [5]. The X-ray reflectivity technique is extensively used to gauge the thickness, density, and surface and interface roughness of ultrathin films [6]. X-ray diffraction (XRD) technique is used to determine the crystallographic structure of thin films as well as bulk materials.
Over the decades, the XRD technique has evolved to characterize different forms of materials—from simple bulk-material to thin films/nanostructures (quantum well, quantum dots, nanorods, complex structures), biological proteins, etc. XRD has also been effectively used to characterize the non-crystalline materials (material exhibiting short-range order of atomic arrangements, i.e., amorphous) such as glasses and liquids. Non-crystalline materials behave as flawed three-dimensional grating structure and yield highly diffused XRD patterns. These diffused XRD patterns are then analyzed using the atomic pair distribution function (PDF) method, which provides crucial structural information like the nearest neighbor distances. Thus, XRD plays a pivotal role in the characterization of both crystalline and non-crystalline solids [7,8,9,10].
Various textbooks and research articles have illustrated the significance of the XRD technique for a better understanding of crystallography, crystal planes, and reciprocal lattice point [7,8,9, 11,12,13,14,15,16]. Giannin et al. [11] discussed the structural evaluation of nanomaterial powder and polymer samples using the XRD technique. Benmore [12] published a review article related to high-energy XRD. Bunaciu et al. [13] have published a critical review of XRD and its application, but they have explained applications, mostly related to chemistry-related samples. Moram et al. [14] have compressively reviewed the high-resolution XRD (HRXRD) study of III-Nitrides epitaxial layers and multilayer device structures. They have only discussed the HRXRD related study of a particular family of samples, i.e. III-Nitrides. Recently (2019), Ameh [15] has discussed the basics of crystallography and XRD; but he has not discussed the experimental aspects of XRD for thin films and bulk samples.
In the present review, we have discussed the applications of different XRD analysis techniques recently used for thin-film samples. This article covers a brief introduction to the concept of the XRD, its instrumentation, and sample preparation for the numerous measurements, and their analysis, including the quantitative and qualitative determination of various crystalline parameters: i.e., the existence of defects in a particular crystal; residual-stress, texture, crystallite size, degree of crystallinity, multiphase content identification in thin films; depth profiling in thin films/multilayered structures, annealing effect of nanostructured thin film by depicting various case studies published recently.
2 Principal and theoretical aspects of X-ray diffraction
Since the German physicist Wilhelm Conrad Röntgen [16] produced and detected X-rays or Röntgen rays in 1895 in his laboratory in Wurzburg, X-rays are used in various fields i.e., medical, physics, chemistry, geology, etc. In the beginning, X-rays were used in medical science. After the discovery of XRD in crystalline materials by Laue and Bragg, this non-destructive technique has been playing a crucial role in determining the structural information of a wide variety of materials [17, 18].
This technique is based on the diffraction of X-rays, whose wavelength is equivalent to the distances between atoms in condensed matter. When X-rays are irradiated onto any crystalline material (material exhibiting a long-range atomic arrangements), which acts as a well-defined three-dimensional grating, a diffraction pattern of definite intense spots is generated. These diffraction spots are popularly known as Von Laue/Bragg diffraction spots.
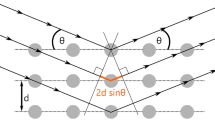
In the year 1915, the father-son duo, Sir W.H. Bragg and Sir W.L. Bragg had received the Nobel prize "for their services in the analysis of crystal structure through X-rays". They discovered the famous Bragg’s law, which correlates the XRD peaks appearing at a certain incidence angle (theta) with the interplanar spacing of the crystal under study:
where dhkl be the interplanar spacing of the (hkl) plane; θB be the Bragg angle; λ be the wavelength of incident X-ray; n is an integer. It was direct evidence for the regular atomic structure of crystals postulated for centuries [10, 19]. Figure 1 represents Bragg’s law for real as well as reciprocal space geometry [14].

Bragg’s law a in real space b in reciprocal space [14]
After growing the crystal; material science researcher uses the XRD characterization method to identify the material phase first. For phase identification material science researcher first uses the Bragg’s law to find out the d spacing and identify the interplanar spacing of the crystalline material. The XRD diffractogram is used to analysis various other structural parameters i.e. particle size, degree of crystallinity, orientation parameter, strains etc. Above mentioned parameters are very important in the material science for further processing of the materials. Details of these parameters extraction can be seen in Sect. 4.
3 Geometries of X-ray diffraction (XRD) systems
X-ray diffraction is a very useful technique, which is used routinely to recognize the crystalline phases present in materials and analyze the materials’ structural properties. XRD system comprises three primary components: (a) the X-ray source (tube), (b) the goniometer (which contains a sample-holder and provide the movement to sample), and (c) the X-ray detector. The X-ray radiation (a mixture of continuous and characteristic spectra) generated from X-ray tube is filtered using foils or monochromator to generate monochromatic X-rays, which are used for XRD from the material under investigations. Usually, copper, molybdenum, and cobalt are commonly used as target materials. Out of them, copper is the most widely used target material for crystal diffraction, with characteristic radiation at 1.5418 Å (popularly known as Cu Kα radiation). The emanating X-rays are then collimated and directed onto the sample. The constructive interference between the scattered X-rays from the constituent planes of atoms gives rise to the diffraction phenomena according to Bragg’s equation [7,8,9,10]. A detector records the intensity of the diffracted beam at different X-ray incident angles. The plot of the diffracted X-ray intensity at different 2θ angle is known as X-ray diffractogram.
Primarily, XRD theory can be classified into two categories based on the kinematical and dynamical theory [20,21,22]. In the kinematical diffraction, the diffraction intensity is directly proportional to the square of the amplitude, and the multiple scattering phenomena are excluded. This theory also neglects the refractive index of the matter in the X-rays wavelengths region (which is less than 1 by a factor in 10–6); therefore, it ignores the fact that the incident beam is refracted to an internal angle slightly smaller than the external one. These factors are considered in dynamical theory; which, takes into accounts multiple scattering, extinction, and the slight deviation of the refractive index from unity [20, 22,23,24]. Commonly, the kinematical theory-based XRD technique is being used in the thin films samples in Grazing incidence XRD. The dynamical diffraction theory is being applied for the analysis of the XRD patterns from the bulk single crystal, epitaxial layer, heterostructures, quantum-well, and super-lattice structures etc.
Figure 2 shows the scattering geometry during the XRD. In the figure, ki denotes the incident wave-vector; ks be the scattered wave-vector; ω is the angle of incidence ray from the surface of the sample; φ be the angle between the lattice plane (hkl) and the sample surface. If φ = 0 i.e. the reflecting lattice planes and the sample surface are parallel then, the Bragg diffraction is termed “symmetric”. Bragg diffraction is termed “asymmetric” when φ ≠ 0. For measuring symmetric Bragg reflection (φ = 0, ω+ = ω− = ω ± θ), a θ − 2θ scan is being used in the traditional powder diffractometer. In the ω scan, the sample is rotated, i.e. ω changes and the detector fixed in position with wide-open entrance slit.

Depending upon the types of materials and the types of information sought, different XRD geometries can be used. A brief about the different geometries are discussed below:
3.1 Powder XRD
The powder X-ray diffraction (PXRD) technique is primarily used for powder, palette, and thick film polycrystalline materials. In this configuration, the sample rotates in the path of the collimated X-ray beam at the incidence angle θ, whereas the detector is mounted on the diffracted arm (situated at an angle of 2θ) to collect the scattered X-rays. A goniometer is used to maintain the incidence angle and rotate the sample accordingly. Powder X-ray diffraction system is based on Bragg Brentano geometry [7,8,9].
In powder geometry, all the three, i.e., source, sample, and detector, are kept on the circumference of a circle known as the focusing circle. Since all three are at the circumference of the circle, the incidence beam focusing of this geometry is extremely good. The schematic diagram is shown in Fig. 3. Here, the circle is known as goniometer circle on which source and detector lie on the circumference and the sample is kept at the center of the circle. In Powder XRD scattering vector (Q) and surface normal (S) are aligned vertically parallel. In front of the detector, a graphite curved crystal monochromator is used. Its divergence is ~ 0.1°.

Adapted from Ref. [25])
Schematic diagram of powder X-ray diffraction (a) using diffractometer circle Q is the scattering vector and S is the surface normal. b Experimental setup (
3.2 Thin-film XRD: grazing incidence X-ray diffraction (GIXRD)
In the thin-film geometry, the specimen should be in the form of a thin-film deposited on to the substrate. In this geometry, especially the X-ray beam enters the specimen with a small angle of incidence from the surface. Due to this variant, this geometry is known as grazing incidence X-ray diffraction (GIXRD). In this case, parallel X-rays are falling on the thin film or multilayer at a very small angle (0.5°–2°), i.e., fixed grazing incidence and diffraction phenomena occur, and the diffracted beam are detected by the detector by scanning along 2θ of the specimen. In this case, the incidence angle is fixed, and the crystallites are randomly oriented; however, the outgoing rays make the diffracted angle (2θ) with the incoming rays. If the scattered X-rays are collected and plotted with 2θ then Braggs’ condition is comparable with the symmetrically measured condition (i.e., powder X-ray diffraction). A fixed incidence angle limits the X-ray penetration into the thin film of the sample while increasing the incidence angle its penetration increases. The penetration depth and information depth in GIXRD can be determined by Eqs. (2) and (3) respectively.
τ is the penetration depth and τα is the information depth from GIXRD geometry. Where α is the angle of incidence, μ is the linear absorption coefficient, and kα is the asymmetrical configuration factor [1/sin (α) − 1/sin (2θ − α)] which depends on GIXRD geometry. Gibaud et al. discussed in detail about both these depths and their impact XRD experiments [26].
GIXRD geometry comprises of a flat crystal graphite monochromator crystal in the diffracted arm and used to place in front of the detector. The resolution of GIXRD geometry is around (0.18–0.25) degree. The schematic diagram of the GIXRD is shown in Fig. 4. Apart from phase analysis of the thin films, the depth profiling of the multilayer can also be done using this geometry. This is the added advantage of this geometry.

adapted from Ref. [25])
Schematic diagram of thin-film grazing incidence X-ray diffraction (a) using diffractometer circle Q is the scattering vector and S is the surface normal which is not parallel for each other. b Experimental setup of GIXRD (
3.3 High resolution X-ray diffraction (HRXRD) [14, 20,21,22]
High-resolution X-ray diffraction is a powerful tool for bulk single crystals, epitaxial layers, heterostructures, and supper lattice structures. Peaks from single-crystal/epitaxial layer materials are sharp (line width is very small) due to very low defect densities of the material, a high angular resolution XRD system is required for the analysis of such samples. Due to diverged rays (Δθ ~ 0.10°) and high wavelength spread (δλ/λ ~ 10–3), the single-axis diffractometer is not able to resolve the individual Bragg peaks of epitaxially grown multilayer structure, because of small mismatch between substrate and epi-layer. Therefore, high-resolution XRD is needed to resolve Bragg peaks for substrate and layers individually. Using the HRXRD technique, one can determine the crystalline quality of bulk single crystal/epitaxial layers, estimation of the epitaxial structure layer composition, the thickness of multilayers, strain status, the absolute lattice constant, and defect densities and probably their nature, etc. This technique is being widely used to determine the relaxation of epilayers and/or multilayer structure by using reciprocal space mapping (RSM). In this review, the details of HRXRD techniques are not discussed. It is explained elsewhere [14,15,16,17,18,19,20,21, 26].
4 Application of XRD/GIXRD techniques
X-ray diffraction techniques are widely used to characterize the material properties of nanostructured thin films and bulk crystal samples. The essential information which can be extracted by using XRD techniques include (i) phase identification, (ii) degree of crystallinity, (iii) structure determination, (iv) determination of lattice parameters, (v) crystallite size and strain determination, (vi) uniform and non-uniform strains, (vii) preferred orientation parameter i.e., texture coefficient, (viii) residual stress, and (ix) depth profiling of multilayer in GIXRD geometry [7,8,9, 26]. The main applications are discussed as follows:
4.1 Phase identification
After growing the thin film, powder, or bulk samples in almost all the branches of science, XRD is one of the quickest methods to get information about the phase formation in the sample. The presence of long-range ordering in the samples will show sharp XRD peaks due to constructive interference of diffracted X-rays; whereas, short-range ordering yield broad hump because of destructive interference start occurring by the scattered beam.
The phase, crystalline quality of any sample in terms of single crystalline, preferred oriented and non-preferred oriented polycrystalline sample are used to determine by indexing the XRD peaks of the sample. Usually, three main techniques are being used for indexing a diffraction pattern, (a) comparison technique (measured XRD pattern is compared with the standard International Crystallographic Diffraction Data (ICDD) files); (b) analytical technique; (c) graphical technique. A comparison of the XRD pattern from ICDD files of three or more intense peaks with their positions and intensity of XRD pattern are used to identify the crystalline phase [7,8,9]. Retwield refinement method is also applied to X-ray powder diffraction, and it is used with the great success of known crystal structure [26,27,28,29]. It is widely used to determine the structural parameters of the powder samples material.
4.2 Degree of crystallinity [29,30,31]
In the mixture of the long and short-range ordering, the crystalline XRD peaks overlap with the amorphous hump. The presence of the crystalline phase in a sample can be determined in terms of the degree of crystallinity (%), which is defined as:
where Σ Itot is the integrated area of the whole X-ray diffractogram; Σ Inet is the integrated area of crystalline XRD peaks (the whole diffractogram has subtracted the hump due to amorphous content), and Σ Iconst.bgr be the constant background from the diffractogram. After plotting, the XRD pattern degree or percentage of crystallinity of the powder/thin film samples can be estimated by the ratio of crystalline XRD peaks integrated area to the total integrated area (XRD pattern).
4.3 Structure determination
Crystals with precise periodicities over long distances have sharp and clear XRD peaks. For a clear XRD profile, a very slow scan speed is preferred. If the XRD pattern is showing the crystalline structure, then by indexing step by step starting from cubic to the monoclinic, one can determine the structure of the sample. After finding out the crystal structure, i.e., cubic, then cubic forms (like simple cubic, body-centered, face-centered, etc.) can be estimated using the selection rule (i.e. XRD peaks at (hkl) plane and (h2 + k2 + l2) value) as mentioned in Refs. [8,9,10]. If the ratio of (h2 + k2 + l2) values are 1, 2, 3, 4, 5, 6, 8, 9 than the crystal is simple cubic. If the ratios are even numbers then the crystal is BCC. The ratio should be 3, 4, 8, 11 for FCC. Retwield refinement analysis of XRD data can also be used for known polycrystalline sample single and multiphase and used to provide the sample structure as well as lattice parameter [26,27,28].
4.4 Lattice parameters
The famous Bond method [32] is an accurate technique for the determination of the absolute lattice parameter of bulk single crystals. Fewster [33] and Fatemi et al. [34] showed the determination of single bulk crystal/epitaxial layers lattice parameter up to four decimal places accuracy by taking appropriate symmetric and asymmetric planes. For polycrystalline crystal, the absolute lattice parameter is used to determine by indexing the XRD peaks for different planes [8]. This procedure is valid when the measurement is done at constant temperature, small step size, large integration time, and appropriate large-angle 2θ XRD peak [33]. To reduce the minimum possible error in 2θ position while scanning in 2θ step size should be as small as possible, and the scanning rate should be as slow as possible [7, 8, 33]. The extrapolation of Nelson–Reley function plot for various reflections of the sample is used to determine the lattice parameter of polycrystalline thin films [7,8,9].
4.5 Crystallite size and strain determination
Nanostructured thin films and nano materials are usually comprise the random distribution of particle sizes. The physical properties of nanomaterials depend on its particle size, defects, and strain in the material. Thus, the characterization of these parameters is very important. The XRD technique can be used to determine the crystallite size and strain of these nano-materials. While calculating the crystallite size using FWHM of the XRD peak, it can exaggerate the exact value since the larger crystallites/grains enhance the intensity, while the smaller grains/crystallite merge in the base of the peak. In 1918, Scherrer suggested the equation (known as Scherrer equation) for crystallite size determination using integral width of the diffraction profile [35]:
where λ be the X-ray wavelength; D is the average particle size; βhkl (Δ2θ) is the FWHM (full width at half maximum) of the reflection peak located at angle 2θ. k be the Scherrer constant which is dimensionless shape factor. The value of k is typically ~ 0.9; but later it was noticed that the magnitude of the Scherrer constant depends on the crystallites shape/geometry. Unless the shape/geometry of the crystallites and distribution are known, there is an uncertainty in k. The value k was found to be varying from 0.62 to 2.08 with the actual shape of the crystallites [26, 36]. This method is applicable to determine the crystallite size is the order of 0.1 μm. Another limitation of Scherrer method that peak broadening includes the stress/strain factor. The use of the Scherrer equation might be a more appropriate method to find the particle size for stress-free samples.
In 1953, Williamson and Hall proposed a method to get the particle size and strain in nanomaterials using the broadening of XRD peaks, this method is now commonly known as the Williamson hall (WH) plot method [37, 38]. This WH plot method is widely used to separate the particle size and strain contribution from the broadening of XRD peaks [35, 39, 40]. Applying this method microstrain and particle size in the sample can be determined using the equation written below:
where λ, β, k are explained above, ɛ be the microstrain present in the sample. Using Eq. (6) \({\beta }_{hkl}\mathrm{cos}\theta\) is plotted with \(\mathrm{sin}\theta\) for a particular diffraction pattern of polycrystalline samples and gets a straight line. The intercept and slope of the straight line will give the value of micro-strain and average particle size. This method is widely used nowadays for polycrystalline thin-film samples for microstrain and particle size analysis.
4.6 Uniform and non-uniform strains
Lattice strain can be explained as the ratio of change in the lattice spacing with the original lattice spacing along a certain direction [7, 8]. In XRD, the peak position is related to average elastic strain within the scattering grains, and the peak shape is associated with the crystallite size and strain (micro-strain), which is depicted by the average spread in lattice parameter values. The peak broadening can be attributed to the anisotropic elastic and plastic properties of each crystallite, which in turn leads to the non-uniform distribution of inter- and intra-grain elastic and plastic strains [41]. If there is a uniform strain in the crystal i.e., the interplanar spacing (d-spacing value) differs from its original value; the XRD peak position will shift from its’ original peak positions. If the sample has a non-uniform strain, several crystallites are strained with one another, the diffraction will occur from all crystallites, causing an increase in FWHM of the XRD peaks.
4.7 Preferred orientation (texture coefficient)
The crystallographic orientation in the nanostructured thin films is not always random and shows some preferred crystallographic orientations. Thus, a strong tendency for the crystallites to be oriented along a particular crystallographic direction is known as preferred orientation. Nanostructured thin films, grown on different substrates, tend to follow the crystallographic orientation of the substrate and thus, may get oriented. The intensities of the XRD peak in the preferred crystallographic orientation should be high as compared to other XRD peaks. In the partially preferred oriented thin film samples, some other XRD peaks may also be appear with relatively higher intensities. The texture coefficient (γ) is used to calculate the preferred orientation parameter by the method proposed by Harris et al. [42]. Later many other researchers are also used the texture coefficient value to find out the orientation parameter for thin films sample [43, 44].
Texture coefficient TC (γ) for the thin films can be determined as follows:
where Ihkl is the intensity measured from XRD pattern and Iohkl is standard intensity taken from ICDD data card for a particular (hkl) planes, and N is the number of diffraction peaks.
4.8 Residual stress determination
If a uniform external load is applied to a rigid body, it deforms elastically. The stress imposed during this process in the body is known as applied stress. If the external load is removed from the body and it will still be in the stressed condition, the remaining stress in the absence of external load in the body is known as residual stress [7, 8]. In natural and artificial materials, residual stress arises from the difference in the thermal expansion coefficient of the material (substrate and layer), known as the thermal stress. If the material deformed elastically, the mismatch in the lattice parameter of the constituents’ grains changes from its relaxed value, such stress is known as mismatch stress. Another stress is the internal stress arises due to defects, impurities, etc., caused by dislocations and point defects in the materials.
In the thin films residual stress significantly influences the characteristics of electronic and sensor devices. The stresses in thin films are classified into two categories: extrinsic stress, i.e. stress due to mismatch in thermal expansion coefficients, and intrinsic stresses, which arises due occupation of foreign atoms in the material.
The deviation of the lattice structure from its equilibrium state as well as microstructure imperfections resulting inelastic deformation of grain boundaries; generate residual stress in the deposited films [41]. XRD is a unique method to estimate the residual stress in the materials (thin film, bulk sample). The rate of change in the interplanar d-spacing in the sample is equal to the lattice strain for a given (hkl) planes in the direction of plane normal. If a particular material is strained, means d-spacing is contracted or elongated within the crystal lattice, and induces a change the interplanar spacing. This change in d-spacing shifts the diffraction pattern. By measuring this shift precisely, the variation in the d spacing can be estimated; as a result, the strain and subsequently stress within the material can be deduced. There are a few methods that are being used to determine the residual stress in the material.
4.8.1 sin2 Ψ method for bulk or thin-film [8, 45,46,47,48]
If the sample is a bulk or thin film, the thickness of the film should be more than a micron. This method can be used to determine the residual stress by the sin2 Ψ method in the sample. In this method, strain (change in the d spacing with the original d spacing ~ ε) varies with the tilt angle (Ψ). The slope of the plot, determined by the d spacing for (hkl) plane versus sin2 Ψ plot; is used to provide the residual stress in the sample. In this method, the appropriate elastic constant and stiffness coefficient values should be used in the calculations. The relation of strain in terms of interplanar spacing, and stress evaluation equation, is given below:
The above equation is used to estimate the residual stress in any chosen crystallographic direction. Since X-rays impinge over a region on the material, several crystallites and grains will contribute to the XRD measurement. Fig. 5 shows the schematically the in plane stress σφ with respect to two principal stress componant σ1 and σ2. The stress value depends on the grain size and used XRD geometry.

Schematic diagram showing the set-up of the XRD-sin2 ψ in-plane stress measurement where σ is residual stress, ψ is the tilt angle, d is the inter-planer spacing of crystallites, φ is the rotation angle [48]
4.8.2 Modified sin2 Ψ method for thin-film
A slight modification has been proposed by Ma et al. [49] for residual stress determination for thin films (thickness less than a micron). In this case, the incidence angle part is also included in stress calculation. The residual stresses equilibrate over atomic dimensions and balance within a grain or even over several crystal dimensions. The residual stress can be estimated by using the Grazing Incidence XRD technique by determining the strain value in the crystal with various tilt values [49, 50].
where ν be the Poisson ratio, E be the young modulus, ψ is the tilt angle (angle between the sample surface and reflecting plane), α is equal to (θ0 − γ), and γ be the grazing incidence angle. The d\(\alpha \psi\) (hkl) and d0 (hkl) are the d-spacings of the (hkl) plane in the strained-film and that in the bulk, respectively. Using Eqs. (10) and (11), the plot strain (ɛ) verses \(\cos ^{2} \alpha \sin ^{2} \psi\) will show a linear behavior. The gradient of the above mentioned linear Eq. (11) is equal \((\frac{1+\nu }{E})\sigma\). Thus the residual stress (σ) of the thin film samples can be estimated from the slope of \(d\alpha \psi (hkl)\) and cos2α sin2ψ and by combining Eqs. (10) and (11).
4.8.3 Biaxial stress and hydrostatic stress by determination of lattice constant
For cubic/non-cubic crystal using HRXRD peak, the absolute lattice constant can be determined. Using the determined lattice constant and elastic constant values, the Poisson ratio, biaxial-stress can be determined. If stressed samples lattice constant expands in all directions, it means that the sample is under hydrostatic strain [51,52,53]. By determining hydrostatic strain and bulk modulus of the material, hydrostatic stress can be determined.
4.9 Depth profiling of multilayer by GIXRD
GIXRD is also being used as depth profiling in thin-film/multilayered samples. By varying the Grazing incidence angle by taking the appropriate plane and its diffracted intensity can be collected as a function of depth from the recorded X-ray diffractogram [54,55,56,57]. Based on GIXRD the diffractogram is collected by a different set of incidence angle, structural information can be estimated at a different depth from the surface of the layers. Therefore apart from phase analysis, depth profiling of the multilayer structure can also be done using GIXRD.
5 Other complementary state of the art structural characterization techniques
XRD provides structural information of nanostructures and thin films averaged over a large area. It is a very quick, easy, non destructive technique, and required less sample preparation time as compared to other structural characterization techniques, like Transmission electron microscopy (TEM), Neutron diffraction, Raman Scattering, Ion beam channeling, etc.
Like the XRD, the neutron diffraction can also be used for structural characterization. Neutron is the charge-less particle and has non-zero magnetic moment. In this experiment, the neutrons interact with the nuclei of the atoms and scattered by magnetic field also [58]. So it gives the crystalline structure as well as magnetic structural information of the material. Using this method sample can be analyzed in hot and cold conditions also. Sometimes neutron diffraction is advantageous for determination of light elements and magnetic ordering of the materials. It can be highly used in the magnetic materials. But due to high cost of neutron generation its use is very limited in day to day experiments. The electron diffraction experiments by TEM are also widely used for materials crystal structure determination. The size of the electron beam ~ sub micrometer to nanometer scale in the electron beam diffraction techniques, so this technique is very useful to get precise, localized and specialized information of the thin films samples. Since, in TEM; transmission phenomena is used therefore a lot of sample preparation method is required before performing the electron diffraction experiments [59]. Another limitation; the nature of electrons are charged particles and it interact with matter by coulombic forces. This means the incident electrons feel the influence of both the positively charged atomic nuclei and the surrounding electrons of the atoms.
Raman scattering can also be used to identify the phase of the materials and compounds, stress/strain, crystalline perfection, layer orientation etc. [29]. In the various scattering geometries Raman active modes also depends on the crystal symmetry. Therefore by indexing the Raman peaks one can identify the phase of the material and orientation of the layer. Broadening of the Raman line is responsible for crystalline perfection of material. Raman peak shift is used for stress/strain estimation of the material. Other important advantage of Raman scattering that it can be used to analyze the ultrathin/2D layered structures also [60]. In spite of several advantages there are some limitations also. It is the indirect technique and sometimes Raman scattered rays doesn’t show any useful scattered signal during the experiments, the reason might be the low grain size of the polycrystalline material and high polar nature of the crystal.
Some other optical characterization method (i.e. optical absorption and photoluminescence (PL)) can also used to determine the crystalline perfection qualitatively. Ion beam channeling can also be used to identify the crystal orientation but generation of high energetic ion beam is a very complicated process, costly, uneasy availability, so it’s use in the day to day sample analysis are not possible.
The details of the information about the techniques and the parameters which we infer are tabulated in Tables 1 and 2.
6 Case studies
6.1 Phase identification, degree of crystallinity, crystallite size and texture coefficient analysis of AlN thin films on Si (100): GIXRD [61,62,63,64,65,66,67]
AlN thin-film form is a potential material and widely studied in the last two decades due to its interesting, exotic, and versatile properties such as Wurtzite structure, wide bandgap, high thermal conductivity, highly stable and piezoelectric nature [61,62,63]. Wurtzite crystals are employed in the area of micro-electro-mechanical system (MEMS) based sensors, and actuators due to their polar structure and piezoelectric characteristics (due to non-centrosymmetry) [64, 65, 68]
The evolution of structural properties and orientation parameter in AlN thin films with the variation of its thickness is studied by Pandey et al. [66, 67]. The films were grown on Si (100) substrates by reactive DC magnetron sputtering technique. Four layers were grown by varying the deposition time with 10 min interval time starting from 25 min (thickness: 300 nm), 35 min (thickness: 430 nm), 45 min (thickness: 630 nm), and 55 min (thickness: 830 nm) named S1, S2, S3, and S4 respectively. The phase analysis of the AlN layers was done using the GIXRD method. In GIXRD, the incidence angle of X-rays ~ 2° was fixed from the surface of the sample and after occurring the diffraction, the diffracted beam was detected by the detector by scanning (2θ angle) from 25° to 75° using Cu Kα radiation.
Figure 6 shows the XRD results of the four AlN layers to be discussed. The AlN layers showed wurtzite structure with XRD peaks along (100), (002), (101), (102), (110), (103) and (112) planes. The diffractogram of sample S1 and S2 showed the peak intensity of (100) plane was relatively high as compared to the (002) peak. Apart from this, the intensities of the other peaks were very small. As the thickness increased (deposition time increased); the orientation of the films along (002) plane was more favored as compared to (100) and it was evident from the increased intensity of (002) peak as compared to other existing planes. This indicates the first sputtered AlN layer growth started in the (100) orientation and after a certain time competitive growth started between the <100> and <001> directions. The peak intensity of the (103) plane diffraction angle 2θ ~ 65.9° (θ = 32.95°) was found to be enhancing in tandem with the (002) XRD peak intensity as shown in Fig. 6. This increase can be explained by the GIXRD geometry. The low incidence angle (α) almost (~ 2°) was used to record XRD pattern. The diffraction angle (θ) for (103) plane θ = 32.95°. In GIXRD geometry the incidence X-ray angle (θ − α) for (103) plane, was ~ 30.95°, this angle was very close to the inter-planer angle (31.63°) between (103) and (002) planes. The incidence X-ray angle of (103) peak is twice the angle of X-ray incidence for (002) plane (θ − α) ~ 16°. As a result, the XRD peak intensity (103) plane of the c-axis preferred oriented film was increased with the increase in (002) plane XRD peak intensity.

XRD pattern of AlN thin films on Si (100) substrate of four samples by the varying film thickness [67]
6.1.1 Degree of crystallinity and crystallite size [30, 31]
The degree of crystallinity was extracted from the XRD data for AlN layers from Fig. 6 as explained in Sect. 4.2. The degree of crystallinity of the AlN films was calculated by the Eq. (4) as explained in the previous Sect. 4.2. It was found that the degree of crystallinity increases with the film thickness. Figure 7a showed the degree of crystallinity for S1, S2, S3 and S4 were 61%, 67%, 92% and 97%, respectively. The crystallite sizes of the AlN layers were determined from the Williamsons hall plot analysis method using Eq. (5) [37, 39, 40]. Figure 7b showed the behavior of crystallite size as a function of AlN film thickness. The average crystallite size increased from 8 to 19 nm as the film thickness increased as shown in Fig. 7b.

a Degree of crystallinity. b Average crystallite size of AlN layers on Si(100) as layer thickness increases. c Texture Coefficient value for XRD peak (100) and (002) of AlN thin films [67]
6.1.2 Orientation parameter (texture coefficient)
The orientation parameter of the thin films can be represented as the texture coefficient (γ), it explains about the orientation of the crystallites in a particular direction [42,43,44]. The texture coefficient (γ) of AlN films was calculated by Eq. (6), explained in Sect. 4.7. Texture coefficient determination for AlN layers from Eq. (6), Io be standard intensity JCPDS data file and N be the number of diffraction peaks from XRD profile. For AlN thin film JCPDS file No. 79-2497 was taken to determine the texture coefficient. The texture coefficient along (100) and (002) plane was determined for AlN samples and shown in Fig. 7c as a function of film thickness.
If the value of the texture coefficient is greater than unity for a particular plane, then the film is preferred along that plane. AlN thin film samples XRD data is compared with the corresponding JCPDS data, it was found that from the figure that the AlN layers of lower thicknesses (300 nm and 430 nm) were oriented along (100) as well as (002), while the AlN films having larger thicknesses S3 (630 nm) and S4 (830 nm) are preferentially oriented along (002) plane (<001> direction). Pandey et al. have explained the reason of orientation transformation in their paper [67]. The Orientation transformation can be explained by the combined effect of strain energy and surface energy films [69, 70]. The surface energy of (002) plane (365 meV/Å2) is as compared to (100) plane (346 meV/Å2) [69]. Due to very close surface energy first growth starts along <100> as well as <001> direction and as thickness increases strain energy along <001) direction dominates. Therefore above the thickness of ~ 430 nm AlN films on Si (100), the strain energy becomes dominant and it changes the crystallites in c-axis preferred oriented. As a result, XRD peak intensity of (002) plane has shown the enhanced value for S3 (630 nm) and S4 (830 nm) AlN layers.
6.2 Residual stress determination of thin films/multilayer samples
6.2.1 Evaluation of residual stress AlN thin films on Si substrate by modified sin2 Ψ method using GIXRD geometry [49, 67]
Pandey et al. demonstrated the residual stress determination of AlN thin films by the modified Sin2 Ψ method using GIXRD geometry [67] as explained in Sect. 4.8. Four AlN thin films deposited on Si(100) by varying thickness as discussed in the previous section S1, S2, S3 and S4 are being used to estimate the residual stress.
In this method higher angle XRD peak with a sufficiently large intense peak has been chosen to reduce the error in strain (change in d-spacing) and hence to minimize the error in residual stress estimation. Using this analogy, the XRD peak corresponding to the (110) plane (at 2θ = 59.8°) was chosen for the stress analysis of S1 and S2 samples; whereas, the XRD peak corresponding to the (103) plane (at 2θ = 65.9°) was chosen for S3 and S4 samples. Around this 2θ peak position, various XRD patterns (range ~ 4°) have been measured at several tilt angles (ψ). Angle (ψ) taken from 0 to 45° at the different equiangular intervals. Figure 8 shows the 2θ scan for (103) plane in GIXRD geometry by varying tilt angles for the sample S2 and S4. One can see as ψ angle increase the intensity of the XRD peak reduces and peak position is shifted in higher angle side. It infers the inter-planer distance might be reduced and clearly indicates the film is under compressive stress in nature. Similar measurements have been done for other samples S1 and S3 also but figure not shown here.

In GIXRD geometry: 2θ scan of a sample S2 for (110) plane. b Sample S4 for (103) plane by varying tilt angle (0°–45°). Plot a to i represents the XRD pattern by varying tilt angle (ψ = 0° to 45°) at every 5° steps
For the quantitative compressive residual stress value determination modified sin2ψ method is used in which d-spacing value was plotted with sin2ψ cos2α as discussed in the previous Sect. 4.8.2. Using this method as explained earlier Sect. 4.8.2, the d spacing and sin2ψ cos2α plot should show the linear behavior. The slope with appropriate elastic constants gives the value of residual stress of AlN thin film.
Figure 9 shows the plot of d-spacing versus sin2ψ cos2α for the (103) plane (in the S3 and S4 samples), and the (110) plane (in the S1 and S2 samples). Residual stress was estimated using Eqs. (10) and (11) by taking the stress-free value do values of (110) plane (1.555 Å) and (103) plane (1.414 Å) respectively. For AlN material, E and ν are taken to be 322 GPa [71] and 0.236, respectively [72]. These values were used to calculate the residual stress of AlN layers. The estimated residual stresses using this technique were found to be—2.1 GPa (S1),—1.5 GPa (S2),—1.1 GPa (S3), and—0.7 GPa (S4), respectively.

Plot of d spacing versus cos2α sin2ψ of four samples a S1, b S2, c S3 and d S4 for residual stress determination by XRD [67]
Thermal mismatch based stress will be the same because all the films were deposited at a fixed temperature (550 °C). The variation of the residual stress value is due to the presence of internal stresses in the films which are due to the variation of microstructure in the films; It might be the probable reason for the change in residual stress in the films. Films microstructures contain grain boundaries, defects, and impurities, etc. The residual stress variation seems to be due to the microstructure of the AlN films and atomic shot peening (due to Ar ion impingement) during the sputtering process. The microstructure FESEM analysis discussed in Ref. [67], at the lower film thickness small grains were observed therefore its grain boundary density (sum of the perimeter of grains per unit surface area) would be large. In comparison as thickness increased, grain boundary density was reduced because of large grain size. From FESEM image as discussed in Refs. [66, 67], As at the initial stage, the probability of impurity incorporation seems to be higher due to large grain boundary density, As a result, films were under high compressive stress at lower thickness. As the thickness increased, the probability of impurity incorporation decreased due to reduced grain boundary density and the larger grain size of thick films as well as sub-grain growth at the grain boundaries. Thus as the AlN layer thickness increased the compressive stress decreased as discussed in the previous paragraph.
6.2.2 Estimation of residual stress in BiFeO3/Pb(ZrTi)O3 multilayer thin film on Si substrate by sin2 Ψ method [73,74,75]
Multilayers of PbZrTiO3 (PZT) and BiFeO3 (BFO) were studied by Dutta et al. [73, 75] as an ‘artificial multiferroic’ material to obtain superior magnetic and ferroelectric properties coupled with low leakage current. However, due to the mismatch in the coefficient of thermal expansion of PZT and BFO layers, residual stress is bound to generate while annealing at an elevated temperature. Dutta et al. have studied residual stress analysis by the XRD method on PZT and BFO multilayers on oxidized Si substrate [74, 75]. The PZT and BFO multilayers were deposited via chemical solution deposition technique on the oxide-coated silicon substrate. To grow crystalline multilayer film on the amorphous surface (SiO2), a ZrO2 seed layer was introduced in between. The total stack of the multilayer is around 170 nm. Crystalline structure and phase of the PZT-BFO multilayer film were analyzed by XRD recorded in GIXRD geometry as shown in Fig. 10. The XRD pattern showed that the deposited ZrO2 seed layer is a polycrystalline monoclinic structure having orientations along (110), (211), (220), (003), and (310) planes. The PZT-BFO multilayer showed polycrystalline perovskite structure having XRD peaks along (100), (110), (111), (200), (210), (211), (220), and (310) planes as shown in XRD pattern (Fig. 10).

X-ray diffractogram of ZrO2 buffer layer and PZT-BFO multilayers [75]
From the XRD pattern in Fig. 10, (200) and (211) planes were chosen for residual stress analysis. Hence, the XRD plot was recorded for the above-mentioned planes (range 4°) different tilt angles ψ (0°, 15.50°, 22.21°, 27.58°, 32.31°, 36.70°, 40.89°, and 45°) as shown in Fig. 11. After the measurement, the d-spacing versus sin2ψ was plotted and the plot was showing the linear characteristics. The variation of d-spacing of (200) and (211) planes with sin2ψ were shown in Fig. 11. Using Eq. (8) the strain (ɛ) value in the crystal was estimated. For strain calculation, the d spacing for thin films was calculated from XRD and d spacing for bulk value was taken theoretically. Using the slope of ɛ and Sin2ψ and by using Eq. (9) the residual stress (σ) value of the sample was estimated. In the present PZT-BFO (3:2) multilayer film, the effective elastic modulus Ef and Poisson’s ratio vf were estimated by weighted average as follows:

XRD peaks of a (200) and b (211) planes at different tilt angle ψ [75]
The residual stress value estimated by XRD was very well matched with the macroscopic stress determined by the based wafer curvature method (Stoney’s equation) [76]. The residual stress of the ZrO2 layer was also estimated by the wafer curvature method and found to be tensile ~ 265 MPa [75, 77]. The thermally grown SiO2 layer on Si possesses compressive stress of 1000 MPa [78]. Thus, the sum residual stress values of PZT-BFO multilayer and ZrO2 would be almost canceling with the SiO2 layer. As a result, a quite favorable integration of PZT-BFO multilayer and Si will be useful for MEMS-based devices.
6.3 Influence of Si substrate orientation on structural properties of AlN thin films [79, 80]
The growth, structural and interfacial properties of the AlN thin films is highly dependent on the substrate orientation (due to its different surface atomic arrangements). Pandey et al. [79] studied these effects, the AlN layers were grown on silicon (100), (110) and (111) substrates by DC reactive magnetron sputtering technique. All the films were deposited in identical growth conditions (with a combination of argon (Ar) and N2 gas flow equally (flow rate ~ 10 sccm) and substrate temperature 550 °C). All the films were deposited simultaneously (deposition time: ~ 55 min). The thickness of the layers was examined by the depth profilometer and confirmed by UV–Vis reflectance fringe width calculation. The structural characterization of AlN thin films (with almost the same thickness ~ 900 ± 50 nm) on Si (100), (110) and (111) substrates by reactive DC magnetron sputtering have been presented in this section.
XRD patterns of the AlN layers on the Si (100), (110) and (111) substrates are shown in Fig. 12a. In Fig. 12a the prominent XRD peaks are (100), (002), (101), (110), (102), (103) and (112) which matches well with the AlN polycrystalline wurtzite structure with pure hexagonal phase [JCPDS file No. 08-0262)]. In the diffraction pattern of the entire three samples intense (002) peak is noticed. It clearly shows the grown layers are preferred oriented along c-direction. In the X-ray diffractogram, the second most intense peak appeared around 66° in corresponding to (103) plane for AlN layers. The appearance of intense (103) peak in XRD pattern was explained in Sect. 6.1.

a XRD patterns of AlN films on Si (100), Si (110) and Si (111) by GIXRD. b X-ray rocking curve of (002) planes of AlN films Si (100), Si (110) and Si (111). c Atomic arrangement of in-plane AlN and Si (100), (110) and (111) [79]
Average crystalline sizes of the AlN layers calculated by Scherrer formula [35] as explained in the Sect. 4.5 were found in the range of 20 to 35 nm. The texture coefficient (γ) of the thin films is determined as explained in the previous section 4.7 using Eq. (7). The calculated texture coefficient (along (002) plane was found to be 3.12, 2.90 and 3.87 corresponding to the AlN layers grown on Si (100), (110) and (111) respectively. The in-plane atomic arrangement of Si (111) and AlN film-oriented along <001> direction is similar thus the AlN film on Si (111) along c direction is highly preferred.
Pandey et al. [79] have also analyzed the crystalline quality of the AlN layers on Si (100), (110) and (111) substrates by X-ray rocking curve analysis in HRXRD geometry as shown in Fig. 12b. X-ray rocking curves were recorded along the (002) plane of the AlN layers. The line width of the Rocking curve FWHM (full-width half maxima) usually depends on the lateral coherence length, local misorientation inside the crystallites, defects (dislocations) [81] etc.; it represents the overall crystalline perfection of the AlN layers [79]. The measurement exhibited the FWHM of 4.2°, 13.5°, and 16.0° corresponding to the AlN films deposited on silicon (111), (100) and 110) substrates, respectively. Since the thicknesses of the films were almost the same, therefore the crystalline perfection of the AlN films depends on AlN and Si substrate interface. From HRXRD results, the AlN/Si (111) is having better crystalline quality as compared to the AlN/Si (100) and AlN/Si (110) films.
Figure 12c shows the in-plane atomic arrangement on to the surface of hexagonal AlN and Si (100), Si (110), Si (111). It is well known that Si (100) and Si (110) substrates have square and rectangular in-plane lattice arrangements while Si (111) surface has a hexagonal lattice arrangement (Fig. 12c). Thus Si (111) plane can provide a matched template for AlN (002) oriented films with minimum stress and high orientation as compared to film grown in Si (100) and Si (110) substrates. Due to matched template AlN layer grown on Si (111) shows the vertical columnar structure (~ 0°) whereas film grown on Si (100) and Si (111) might be slanted (12° and 22° slanted) columnar structures (cross-sectional FE-SEM image (shown in Ref. [79]). Thus the substrate orientation plays an important role in the AlN films structure properties which was analyzed by XRD.
6.4 Doping effect in structural properties of BFO (BiFe1−xNixO3) [82, 83]
Researchers have shown significant attention in the growth, characterization, and device fabrication using (BFO) thin-film materials due to its important and useful application in sensors and electronics [84, 85]. In spite of remarkable properties i.e. structural and magnetic, the application of BFO thin films deteriorates due to its low remnant polarization and large leakage current behaviour. This problem may be overcome by fabricating the artificial multiferroic material by growing the BiFeO3 {perovskite (ABO3)} thin films by intermixing the constituent elements in place of B atoms in a significant manner so that the large change in the structure of the material should not occur. Using the site engineering substitution method of divalent alkali metals in BFO the structural, electrical and mechanical properties of materials change enormously. Sharma et al. explained the change in the structural properties by doping the Ni and Ti atoms in BFO on Si substrate during the growth [82, 83].
The effect of Ni doping in BFO on its structural properties was studied in detail by using the XRD technique. The BiFe1−xNixO3 (x = 0, 0.03, 0.05, 0.07, 0.1 and 0.2) films were deposited by spin-coating technique on platinised silicon substrates and subsequently annealed at 550 °C for 30 min. Figure 13a shows the X-ray diffractogram of the BiFe1−xNixO3 thin films on the platinised-Si substrate. The prominent XRD peaks at 23°, 32°, 39°, 46°, 52°, and 57° corresponding to the (012), (110), (202), (024), (116)/112, (214/300) planes matched quite well with JCPDS card No. 71-2494. It confirms the films were polycrystalline perovskite structure having rhombohedral (space group: R3c) phase. The XRD diffractogram does not showed any NiO peak [82, 86].

a XRD pattern of BiFe1−xNixO3 (x = 0, 0.03, 0.05, 0.07, 0.1 and 0.2) films prepared by spin coating inset shows the intensity and peak position variation of (012) and (110) XRD peak. b Determined strain using (012) and (110) plane, c lattice strain in c and a direction of BFO films as increasing the Ni content in BFO layers [82]
In the XRD pattern, it was found that the peak intensity of (012) was increased with the increasing the Ni content in the films; whereas, the (110) plane peak intensity was found to be decreasing with the increase of Ni content in the BiFe1−xNixO3 films (inset of Fig. 13a). Also, the peak positions (012) and (110) were found to be shifted slowly towards the lower diffraction angle (2θ) side as the doping of the Ni was increased. It indicates that the layer was under tensile strain as Ni was introduced in the layers. The reason may be the Ni4+ ions (ionic radius: 0.69 Å) were introducing and occupying their site in place of Fe3+ ions (ionic radius: 0.64 Å). Since the ionic radii of Ni is large as compared to Fe, therefore, it introduces the tensile strain in the perovskite lattice as indicate in the slight shift of (012) and (110) plane XRD peak positions. Figure 13b shows the evaluated tensile strain in the (012) and (110) planes with the increase in Ni doping in BiFe1−xNixO3 lattice.
The effect Ni doping at the Fe site in BFO lattice was analyzed by Rietveld refinement method. The Rietveld refinement method will be discussed later in this review. The estimated lattice parameter of the un-doped BFO layer was around a = 5.5678 Å and c = 13.8411 Å at room temperature by Rietveld refinement analysis. While introducing the Ni content in BFO layer, it was found out the value of a parameter was increasing; whereas, c value was found to be decreasing with the Ni doping in BFO. Figure 13c shows the rate of change of lattice parameters (in terms of lattice strain) along with a (Δa/a) and c directions (Δc/c) as a function of Ni doping. The lattice parameters of the BiFe1−xNixO3 films are reduced up to 1.15% in c direction and increased up to 0.52% in a direction as increasing the Ni content. Thus, the biaxial strain is created in BFO lattice as the Ni substitution occurred in the BFO layers.
Doping the Ni content in BFO films increased the mechanical strength in the layers as explained in the ref [82]. The Elastic modulus of the BFO films was found to be increasing linearly from 51 GPa (pristine) to 80 GPa (20 at.% Ni) while Hardness was also increased (7.5–14.5 GPa) as Ni content was introduced in BFO [82]. The reason for increasing the mechanical strength might be due to the combined effect of biaxial tensile strain and increase in the grain size by incorporation of high elastic modulus material like Ni in BFO lattice.
6.5 Depth profiling by GIXRD
In the GIXRD measurement by varying the incidence angle from (0.2°–5°) the information depth will also vary while collecting the diffracted X-ray intensity from the detector. Using this analogy, apart from structural analysis depth profiling of the thin films/multilayers can also be done just by taking the relative intensity ratio of the detected XRD peak of a particular (hkl) plane.
Many researchers used the depth profiling of the multilayer thin film by varying the grazing incidence angle using GIXRD [54,55,56,57, 87, 88]. Colombi et al. prepared a model for depth profiling of the thin films [53, 54]. Nauer et al. [88] had taken the multilayer and explained the depth profiling by GIXRD.
Nauer et al. [88] explained the depth profiling using the GIXRD method of hetero-junction photovoltaic solar cell structure. Photovoltaic solar cell structure was prepared by electro-deposition of II–VI semiconductor thin films deposited on a transparent conductor/SnO2/Glass template. The structures consist of a wide bandgap window zinc selenide (ZnSe), a light absorber (CdTe), and back contact an ohmic material, the whole structure as shown in the inset of Fig. 14. X-ray diffractogram with varying incidence angle as shown in Fig. 14. Thin layers CdTe, ZnSe and SnO2 were well crystallized as seen from the XRD pattern. As angle of incidence was decreased step by step the entire XRD signal from the multilayers becomes weaker due to the reduction of intensity per unit area as shown in the XRD pattern of hetero-structures (Fig. 14).

X-ray diffractogram by changing the incidence angle [88]
Figure 15 shows the relative intensity plot of CdTe (111), ZnTe (111) and SnO2 (110) by increasing the grazing incidence angle (from 0.2° to 3.0°). The integrated intensity of normalized signals for a particular peak was drawn by decreasing the angle of incidence with the normalized peak intensity. The normalized intensity of CdTe layer (111) was first increasing from very low angles of incidence as expected and finally reached a plateau when the path length of the light through the layer leads to practically complete absorption of the radiation. The diffraction intensity was also simulated (for constant irradiation intensity) by the relation as follows:

a Effect of intensity while changing the angles of incidence. b Depth profiling using GIXRD [88]
In the above relation, the thickness of the CdTe layer represented as d and absorption coefficient (μ) = 1152 cm−1 of CdTe for X-ray wavelength (0.154 nm) had been taken. Equation (14) was considered the fact that the penetration of X-rays in the thin films is limited with the film thickness whenever it reaches the limit of absorption as angle increases. The simulated curves by varying thickness as indicated in the graph were shown in Fig. 15b. The experimental data with normalized intensity was fitted with simulated curves for a thickness, between 250 and 350 nm. The best-fitted value was found for CdTe (320 ± 30 nm). The density of the electrodeposited film was assumed to be less than 100%. Thus the density and surface roughness account for the limited accuracy of the film thickness around 30 nm for the CdTe films.
Thus this case study clearly explains that apart from phase analysis the GIXRD can also be used as depth profiling of the multilayers. It widely utilized to identify the thickness (depth) of the individual layer in the multilayer structures. Bouroushian et al., It was also explained the depth profiling of TiO2 and Ti using low angle incidence GIXRD technique [89].
6.6 Texture analysis of thin-film using XRD by pole figure
The degree of preferred orientation in thin films influences on the properties of the thin film-based devices. After the material growth, grains/crystallites usually not randomly oriented with respect to each other. To examine orientation in a preferred direction XRD might be an ideal tool [42, 90]. Electron microscopy is another tool but it inform about a very small area ~ microns, while XRD allows the information of large area/volume. The XRD peak intensity of preferred orientated thin films changes the intensity distribution of the films as compared to bulk crystal. The texture coefficient value can be used to quantify such distributions in the oriented samples [90,91,92]. A pole figures analysis can be used to study the complete picture of the orientation distribution. Pole figure analysis is also used to find out the nanorod/nano-wire orientation of GaN nano-rods and InAs nanowires grown by the MBE [91, 92].
In this review, the XRD study of preferred oriented growth of ZnO thin film analysis has been chosen as a case study using pole figure analysis [93]. Texture-controlled growth of ZnO films on Si substrates at room temperature by PLD technique had been demonstrated by Hong et al. [93]. Two ZnO films on Si substrate were grown by PLD technique at room temperature by varying the fluencies to get different film orientations. ZnO thin film on Si substrate with preferred orientation of (001), (100) was compared with the powder data by Hong et al. [93]. Pole figure study for (100) and (100) oriented films was done by Hong et al. [93] and explained as follow:
Figure 16a–d show the pole figure for measuring the degree of texture. Figure 16a, b shows the pole figures along [001] and [101] direction of <001> textured ZnO film. Figure 16a shows the one strong peak at the center of the pole figure along the direction of normal of the substrate i.e. (001) pole. The width of the peak (i.e. broadening along the tilt direction) ~ 14°, which means grains are oriented along c direction and aligned from the film surface normal around ± 7°. Although the crystallites are aligned in c-axis but randomly distributed along a-axis in the film-surface, this phenomenon is shown by (101) pole figures as the ring pattern (Fig. 16b). Figure 16c, d are [100] and [101] pole figures from the (100) textured film. The (100) texture layer shows the peak at the center of the pole figure as shown in Fig. 16c. A weaker ring pattern at 60° tilt angle (ψ) is from the symmetrically equivalent <110> direction in the hexagonal crystal structure. The peak width of the (100) pole was measured around ~ 9°, which is showing lower value as compared to the width of the (001) pole. Figure 16d shows the two ring patterns in the pole figure along [101]. The two ring pattern represent <101> and <111>. It represents the randomly distributed crystallites along (101) and (111) within the film plane As a result it represents a ring shape in the pole figure.

ZnO films grown on Si substrates Pole figures of a [001], b [101] poles of the (001) textured film, and c [100], d [101] poles of the (100) textured film [93]
The preferred orientation changes as the surface energy of the film surface changes. The order of texture changed from (001) to (110) to (100), as the order of the atomic planes varied with increasing surface energy which was governed by laser fluence in PLD growth. Therefore this study concludes for a preferred oriented textured film can also be analyzed with great accuracy by XRD pole figure analysis.
6.7 Rietveld refinement analysis method of X-ray diffraction pattern
Rietveld refinement method, used for the analysis of XRD pattern, is an iterative calculation technique described by pioneer Hugo Rietveld for use in the structural characterization of crystalline materials [27, 28]. Rietveld refinement method emphasized to make an XRD pattern theoretically by using the positional parameters of the atoms in the unit cell and cell parameters a, b, c and α, β, γ. The calculated diffraction pattern of the particular material can be fitted by the experimental diffraction pattern using the peak position, peak intensity, background with a multi-parameter mathematical model. The best set of above-mentioned parameters is determined by an iterative calculation procedure. This iterative procedure is being used to minimize the difference between the measured XRD patterns and calculated diffraction profile. While fitting the criterion for goodness-of-fit is the minimization of R and which is proportional to χ2 (statistical test) as follows:
In the above equation M be the number of data points in the diffraction pattern, and Nex and Nc are the numbers of counts in the experimental and calculated data point i.
The crystal structure, lattice parameters, crystallographic site occupancies, peak intensity and sample positioning error are the main refinement parameters. Rietveld refinement method of XRD pattern is widely used for bulk samples crystalline phase and structure analysis [94, 95]. In next section, thin films are explained as a case study for determining the composition using the Rietveld refinement method of the XRD pattern.
6.8 Multiphase identification of TiO2–Cr2O3 nanocomposite films [96,97,98,99]
Transition metal-oxide thin film is being widely used in diverse applications. Researchers used titanium oxide (TiO2) and chromium oxide separately for sensing and capacitive based electronic application [96, 97]. Few researchers in the recent past combined the TiO2 and Cr2O3 and used Ti and Cr oxide composites to take advantage for numerous applications [96,97,98].
Dutta et al. had successfully grown TiO2–Cr2O3 nano-composite films, and evaluated the TiO2 and Cr2O3 composition from the samples by XRD (using Rietveld refinement) and Raman spectroscopy [99]. Thin films of varying compositions of TiO2 and Cr2O3 layers were deposited on platinum-coated silicon (001) template from titanium and chromium targets (4-in.-diameter; 99.6% pure) using a DC magnetron sputtering system. Six samples Cr6Ti0, Cr5Ti1, Cr4Ti2, Cr3Ti3, Cr2Ti4, Cr1Ti5 and Cr0Ti6 of 30 nm thicknesses were grown. Annealing of the samples was done at 800 °C for 30 min in an oxygen atmosphere (O2 flow rate ~ 100 sccm) in a three-zone furnace.
After annealing TiO2 and Cr2O3 phases of nanocomposites were confirmed by XRD analysis. Cr0Ti6 samples peaks were very well matched with the tetragonal rutile phase of TiO2 (JCPDS no. 88-1172) and the peaks corresponding to the Ti0Cr6 sample was found to be matched well with the rhombohedral eskolaite phase of Cr2O3 (JCPDS no. 84-1616) [99]. Other samples were showing the mixed phase of rutile and escolite phases.
To estimate the contents of the composition of the TiO2 and Cr2O3 phases in the nano-composite films, XRD pattern was analyzed by the Rietveld refinement method by using PANalytical High score plus software in multiphase mode. Rietveld analyses of the ultrathin films are shown in Fig. 17a–g. Figure 17h shows the determined content of TiO2 and Cr2O3 phases (%) in the films as a columnar chart.

Rietveld refinement analysis of XRD pattern of the Ti–Cr nanocomposite oxide ultrathin films: a Cr6, b Cr5Ti1, c Cr4Ti2, d Cr3Ti3, e Cr2Ti4, f Cr1Ti5, g Ti6 and h compositions [99]
The Ti6 and Cr6 and samples are found to be pure (100%) rutile and eskolaite phase, respectively. The analysis of the diffraction peaks of remaining samples Cr5Ti1, Cr4Ti2, Cr3Ti3, Cr2Ti4, Cr1Ti5 reveals the mixed amount of rutile TiO2 and eskolaite Cr2O3 phases. From Rietveld analysis, the Cr5Ti1, Cr4Ti2, Cr3Ti3, Cr2Ti4, and Cr1Ti5 samples are found to possess 69.5%, 46.9%, 26.1%, 14.2% and 7.1% of Cr2O3 phase respectively as shown in Fig. 17a–h. Below the Rietveld refinement fitting figure, a different theoretical and experimental plot is shown which is a noisy straight line with a small kink at some positions this may be due to the analysis is done in the nanocomposite thin films. Thus Rietveld refinement method is used to distinguish the composition of the thin film in the nanocomposite TiO2 and Cr2O3 phase. This analysis is very useful to distinguish the formation of nano-composite phases. The TiO2–Cr2O3 nano-composite might be useful in various gas sensing applications.
6.9 Absolute lattice constant determination
To determinate of the lattice constant of polycrystalline samples, XRD pattern should be recorded with very slow scan so that peak position can be measured with great accuracy. In XRD pattern for range of 2θ angles the diffraction peaks were measured, and by using with Bragg’s law and diffraction peak, the interplanar spacing of the sample can be calculated.
Using an X-ray powder diffractometer, lattice parameters can be measured up to fourth decimal accuracy. This accuracy cannot be achieved by taking one peak in diffraction profile to calculate the parameters by using Bragg’s law. Thus several peak positions of XRD pattern should be taken precisely due to specimen surface irregularities, and the difference in the X-ray penetration depth. Nelson and Riley [7, 8] performed an experimental study of a diffractometer who suggested that an extrapolation procedure can be used by taking the several XRD peak position for lattice parameter determination. The first lattice parameter can be calculated by Bragg’s law for {hkl} planes than the lattice parameter can be plotted by Nelson–Riley function (cos2θ/sin θ), after that lattice parameter, is extrapolated to the ordinate. The extrapolated ordinate value is the precise lattice constant value of a polycrystalline sample. Nelson and Riley use the empirical relationship as follows:
θ be the diffraction angle. Nelson–Riley plots for two same samples of different thickness show different plots because their effective centers of diffraction are different. But two extrapolations give nearly the same value of lattice parameter.
In 2013 Mehdikhani et al. used the Nelson–Riley plots and obtained the lattice parameter of the tantalum carbide (TaC) powder phase after 3, 6 and hours milling times [100]. Khemiri et al. has also used the Nelson–Riley extrapolation method to determine the lattice parameter of CuInS2 powder for solar cell application [101]. Figure 18a shows the XRD pattern of CuInS2 and Fig. 18b shows the Nelson–Riley plots using the XRD pattern for different reflections of CuInS2 sample to estimate absolute lattice parameters.

a XRD pattern of CuInS2 sample and b Nelson–Riley extrapolations for CuInS2 sample for lattice constant a determination [101]
Fatemi et al. had shown the accurate lattice constant determination method with the HRXRD rocking curve measurement of the single crystal [31]. In which X-ray rocking curve measurement has been recorded at 0° and by rotating the sample 180° in azimuth direction in both the symmetric and asymmetric scans. By taking the average value, the instrumental offset has been estimated. Keeping the instrumental offset in Bragg’s angle and Bragg’s equation; accurate lattice constant can be to determine accurately. Thus one can say using HRXRD accurate lattice constant can be determined up to five decimals accurately.
7 Future prospects of XRD techniques
Ever since the discovery of the XRD technique, the materials scientists are being continuously using the XRD characterization for thin films and multilayer structures. With the tremendous progress in modern day materials science in multiferroics (BiFeO3, h-YMnO3, CuO etc.); wide band gap materials (i.e. ZnTe, Ga2O3, SiC polytypes etc.); quantum structures (superlattice, quantum dot etc.); topological insulators (Bi2Se3, Bi2Te3 etc.), the importance of XRD is ever growing to extract various structural parameters of these materials. Presence of the residual stress in these state of the art materials also play a pivotal role in determining their final out-come. By using the XRD technique one can estimate the residual stress in these materials.
Since, majority of the semiconductor materials are anisotropic in nature, their optical and electronic properties greatly influenced by the crystallographic orientation of the semiconducting films. Therefore, determination of crystalline orientation is very crucial for modern materials i.e. (1) bulk crystals (SiC, GaN, AlN etc.), and (2) non liner crystal (i.e. lithium niobate (LiNbO3), lithium tantalate (LiTaO3), ZnTe, GaP, etc.). So, XRD pole figure can be used for the orientation determination of the crystal and it will be very useful for the futuristic high power/frequency electronics. Precise orientation cutting in the proper orientated direction of the crystals are very important non-liner crystal which can be done after getting the orientation information by XRD pole figure analysis. Nonlinear crystals can widely be used for terahertz and electro-optic device applications respectively [102, 103].
HRXRD is being highly utilized for structural analysis for epitaxial multilayer heterostructures in the past. But HRXRD can be used more extensively for composition and interface quality, RSMs etc. analysis on various epitaxial structures (i.e. III-Nitride based heterostructures like InGaN/GaN and AlGaN/GaN; InGaAs/GaAs heterostructures etc.) for optoelectronic and high power/frequency electronic device for defence and civil applications.
8 Summary and conclusion
XRD is an important technique, providing quick and comprehensive structural information about the bulk, thin films, nanomaterials and multilayer device structures. This report discussed about the importance of XRD techniques in the characterization of polycrystalline thin films. XRD/GIXRD is extensively used for phase analysis, residual stress, particle size determination, degree of crystallinity, strain, texture coefficient etc. The added advantage of GIXRD is the depth profiling of thin films and multilayer structures. The principles of various methods and their experimental geometries are discussed to understand the fundamentals of XRD measurements. The case studies are discussed to explain the different principles in respective data analysis. Thus, this comprehensive review will serve as a reference for researchers working in the domain. The XRD is an evergreen tool for characterization of materials and thin films of past, present and future.
References
M. Ohring, The Materials Science of Thin Film, 2nd edn. (Academic Press, Boston, 1992).
S.C. Tjong, H. Chen, Nanocrystalline materials and coatings. Mater. Sci. Eng. R 45, 1–88 (2004). https://doi.org/10.1016/j.mser.2004.07.001
P. Muralt, Ferroelectric thin films for micro-sensors and actuators: a review. J. Micromech. Microeng. 10, 136–146 (2000). https://doi.org/10.1088/0960-1317/10/2/307
S.M. Sze, Semiconductor Devices: Physics and Technology, 2nd edn. (Wiley India Pvt, Chichester, 2008).
F.A. Vittoria, M. Endrizzi, P.C. Diemoz, A. Zamir, U.H. Wagner, C. Rau, I.K. Robinson, A. Olivo, X-ray absorption, phase and dark-field tomography through a beam tracking approach. Sci. Rep. 5, 16318 (2015). https://doi.org/10.1038/srep16318
A. Gibaud, S. Hazra, X-ray reflectivity and diffuse scattering. Curr. Sci. 78(12), 1467–1477 (2000)
B.D. Cullity, S.R. Stock, Elements of X-Ray Diffraction, 3rd edn. (Prentice-Hall, Englewood Cliffs, 2001).
C. Suryanarayana, X-Ray Diffraction: A Practical Approach (Springer, M. Grant Norton, 1998).
L.V. Azaroff, Elements of X-Ray Crystallography (McGraw-Hill, New York, 1968).
C. Kittel, Introduction to Solid State Physics, 5th edn. (Wiley, Chichester, 1976).
C. Giannini, M. Ladisa, D. Altamura, D. Siliqi, T. Sibillano, L. De Caro, X-ray diffraction: a powerful technique for the multiple-length-scale structural analysis of nanomaterials. Crystals 10, 1–22 (2016). https://doi.org/10.3390/cryst6080087
C.J. Benmore, A review of high-energy X-ray diffraction from glasses and liquids. Intl. Sch. Res. Notices (2012). https://doi.org/10.5402/2012/852905
A.A. Bunaciu, E. GabrielaUdriştioiu, H.Y. Aboul-Enein, X-ray diffraction: instrumentation and applications. Crit. Rev. Anal. Chem. 45, 289–299 (2015). https://doi.org/10.1080/10408347.2014.949616
M.A. Moram, M.E. Vickers, X-ray diffraction of III-nitrides. Rep. Prog. Phys. 72, 036502 (2009)
E.S. Ameh, A review of basic crystallography and X-ray diffraction applications. Int. J. Adv. Manuf. Technol. 105, 3289–3302 (2019). https://doi.org/10.1007/s00170-019-04508-1
R Novelize, Squire's Fundamentals of Radiology. Harvard University Press. 5th edition. ISBN 0-674-83339-2, p. 1 (1997).
W. Friedrich, P. Knipping, M. von Laue, Interferenz-Erscheinungen bei R€ ontgenstrahlen. Sitzungsber. Math.-Phys. Classe K€oniglich Bayerischen Akad. Wiss. M€unchen. 303–322 (1912).
M. Eckert, Ann. Phys. (Berlin), 524 (5), A83–A85 (2012) https://doi.org/10.1002/andp.201200724.
W.H. Bragg, W.L. Bragg, The reflexion of X-rays by crystals. Proc. R. Soc. Lond. A. 88(605), 428–438 (1913)
D.K. Bowen, B.K. Tanner, High-Resolution X-Ray Diffractometry and Topography (CRC Press, Boca Raton, 2005).
G. Bauer, Optical Characterization of Epitaxial Semiconductor Layers (Springer, Berlin, 1996).
P. F. Fewster, X-Ray Scattering from Semiconductors, Second Edition Imperial College Press, (2003).
D. Taupin, Bull. Soc. Fr. Mineral. Cristallogr. 87, 469 (1964)
S. Takagi, J. Phys. Soc. Jpn. 26, 1239 (1969)
PANalytical X-Pert Pro -MRD HRXRD manual
M. Birkholz, Thin films analysis by X-Ray scattering, Wiley-VCH GmbH & Co. 2006.
H.M. Rietveld, Line profiles of neutron powder diffraction peaks for structure refinement. Acta Crystallogr. 22, 151 (1967). https://doi.org/10.1107/S0365110X67000234
H.M. Rietveld, A profile refinement method for nuclear and magnetic structure. J. Appl. Cryst. 2, 65–71 (1969). https://doi.org/10.1107/S0021889869006558
X. Orlhac, C. Fillet, P. Deniard, A.M. Dulac, R. Brec, Determination of the crystallized fractions of a largely amorphous multiphase material by the Rietveld method. J. Appl. Cryst. 34, 114–118 (2001). https://doi.org/10.1107/S0021889800017908
S.M. Ohlberg, D.W. Strickler, Determination of percent crystallinity of partly devitrified glass by X-ray diffraction. J. Am. Ceram. Soc. 45, 170–171 (1962). https://doi.org/10.1111/j.1151-2916.1962.tb11114.x
A.S. Goikhman, V.M. Irklei, O.S. Vavrinyuk, V.I. Pirogov, X-ray diffraction determination of the degree of crystallinity of cellulose using a computer. Fibre Chem. 24, 80–85 (1992). https://doi.org/10.1007/BF00557189
W.L. Bond, Precision lattice constant determination. Acta Cryst. 13, 814 (1960). https://doi.org/10.1107/S0365110X60001941
P.F. Fewster, Absolute lattice parameter measurement. J. Mater. Sci.: Mater. Electron. 10, 175–183 (1999). https://doi.org/10.1023/A:1008935709977
M. Fatemi, Absolute measurement of lattice parameter in single crystals and epitaxic layers on a double-crystal X-ray diffractometer. Acta Crystallogr. A 61, 301–313 (2005). https://doi.org/10.1107/S0108767305004496
P. Scherrer, Göttinger Nachrichten Gesell. 2, 98 (1918)
J.I. Langford, A.J.C. Wilson, Seherrer after sixty years: a survey and some new results in the determination of crystallite size. J. Appl. Cryst. 11, 102–113 (1978). https://doi.org/10.1107/S0021889878012844
G.K. Williamson, W.H. Hall, X-ray line broadening from filed aluminium and wolfram. Acta Metall. 1, 22–31 (1953). https://doi.org/10.1016/0001-6160(53)90006-6
B.E. Warren, B.L. Averbach, J. Appl. Phys. 21, 595 (1950). https://doi.org/10.1063/1.1699713
V. Mote, Y. Purushotham, B. Dole, Williamson-Hall analysis in estimation of lattice strain in nanometer-sized ZnO particles. J. Theor. Appl. Phys. 6, 6 (2012). https://doi.org/10.1186/2251-7235-6-6
A. K. Singh (ed.), Advanced X-ray Techniques in Research and Industries, Ios Pr Inc, (2005). ISBN 1586035371.
P.J. Withers, H.K.D.H. Bhadeshia, Residual stress. Part 1: measurement techniques. Mater. Sci. Technol. 17, 355–365 (2001). https://doi.org/10.1179/026708301101509980
G.B. Harris, Quantitative measurement of preferred orientation in rolled uranium bars. Phil. Mag. Series 43, 336 (1952). https://doi.org/10.1080/14786440108520972
M. Kumar, A. Kumar, A.C. Abhyankar, Influence of texture coefficient on surface morphology and sensing properties of W-doped nanocrystalline tin oxide thin films. ACS Appl. Mater. Interfaces. 7, 3571–3580 (2015). https://doi.org/10.1021/am507397z
Y. Wang, W. Tang, L. Zhang, Crystalline size effects on texture coefficient, electrical and optical properties of sputter-deposited Ga-doped ZnO thin films. J. Mater. Sci. Technol. 31(2), 175–181 (2015). https://doi.org/10.1016/j.jmst.2014.11.009
I. C. Noyan, J. B. Cohen, Residual Stress, Measurement by Diffraction and Interpretation (Springer-Verlag, New York, 1987). https://doi.org/10.1007/978-1-4613-9570-6
S. K. Gupta, M.G. Gartley, International Centre for Diffraction Data (ICDD), the Denver X-ray Conference (DXC) 52 (1999).
J.N. Reddy, Mechanics of Laminated Composite Plates (CRC Press, Boca Raton, 1997).
Q. Luo, A.H. Jones, High-precision determination of residual stress of polycrystalline coatings using optimised XRD-sin2ψ technique. Surf. Coat. Tech. 205, 1403–1408 (2010). https://doi.org/10.1016/j.surfcoat.2010.07.108
C.H. Ma, J.H. Huang, H. Chen, Residual stress measurement in textured thin film by grazing-incidence X-ray diffraction. Thin Solid Films 418, 73–78 (2002). https://doi.org/10.1016/S0040-6090(02)00680-6
Y. Xi, K. Gao, X. Pang, H. Yang, X. Xiong, H. Li, A.A. Volinsky, Film thickness effect on texture and residual stress sign transition in sputtered TiN thin films. Ceram. Int. 43, 11992–11997 (2017). https://doi.org/10.1016/j.ceramint.2017.06.050
M.K. Ozturk, E. Arslan, I. Kars, S. Ozcelik, E. Ozbay, Strain analysis of the GaN epitaxial layers grown on nitridated Si (111) substrate by metal organic chemical vapor deposition. Mater. Sci. Semicond. Process 16, 83–88 (2013). https://doi.org/10.1016/j.mssp.2012.06.013
S. Chowdhury, D. Biswas, Impact of varying buffer thickness generated strain and threading dislocations on the formation of plasma assisted MBE grown ultra-thin AlGaN/GaN heterostructure on silicon. AIP Adv. 5, 057149 (2015). https://doi.org/10.1063/1.4921757
A. Pandey, R. Raman, S. Dalal, D. Kaur, A.K. Kapoor, Structural and optical characteristics investigations in oxygen ion implanted GaN epitaxial layers. Mater. Sci. Semicond. Process. 107, 104833 (2020). https://doi.org/10.1016/j.mssp.2019.104833
W.C. Marra, P. Eisenberger, A.Y. Cho, X-ray total-external-reflection–Bragg diffraction: a structural study of the GaAs-Al interface. J. Appl. Phys. 50, 6927 (1979). https://doi.org/10.1063/1.325845
P. Colombi, P. Zanola, E. Bontempi, R. Roberti, Glancing-incidence X-ray diffraction for depth profiling of polycrystalline layers. J. Appl. Crystall. 10, 176–179 (2006). https://doi.org/10.1107/S0021889805042779
P. Colombi, P. Zanola, E. Bontempi, L.E. Depero, Modeling of glancing incidence X-ray for depth profiling of thin layers. Spectrochim. Acta Part B 62, 554–557 (2007). https://doi.org/10.1016/j.sab.2007.02.012
C.A.Ã. Kaufmann, R. Caballero, T. Unold, R. Hesse, R. Klenk, S. Schorr, M. Nichterwitz, H. Schock, Depth profiling of Cu (In, Ga)Se2 thin films grown at low temperatures. Solar Energy Mater Solar Cell 93, 859–863 (2009). https://doi.org/10.1016/j.solmat.2008.10.009
X. Jin, Neutron Diffraction Principles, Instrumentation and Application (Nova Science Publishers, Inc., New York, 2013).
D.B. Williams, C.B. Carter, Transmission Electron Microscopy: A Textbook for Materials Science (Springer, New York, 1996).
X. Cong, X.L. Liu, M.L. Lin, P.H. Tan, Application of Raman spectroscopy to probe fundamental properties of two-dimensional materials. NPJ 2D Mater. Appl. 4, 1–12 (2020). https://doi.org/10.1038/s41699-020-0140-4
B. Mednikarov, G. Spasov, T. Babeva, Aluminum nitride layers prepared by DC/RF magnetron sputtering. J. Optoelectron. Adv. Mater. 7, 1421–1427 (2005)
D. De-Faoite, D.J. Browne, F.R. Chang-Dıaz, K.T. Stanton, A review of the processing, composition, and temperature-dependent mechanical and thermal properties of dielectric technical ceramics. J. Mater. Sci. 47, 4211–4235 (2012). https://doi.org/10.1007/s10853-011-6140-1
C. Caliendo, P. Imperaton, E. Cianci, Structural, morphological and acoustic properties of AlN thick films sputtered on Si(001) and Si(111) substrates at low temperature. Thin Solid Films 441, 32–37 (2003). https://doi.org/10.1016/S0040-6090(03)00911-8
K. Tonisch, V. Cimalla, C. Foerster, H. Romanus, O. Ambacher, D. Dontsov, Piezoelectric properties of polycrystalline AlN thin films for MEMS application. Sens. Actuators A 132, 658–663 (2006). https://doi.org/10.1016/j.sna.2006.03.001
A. Ababneh, M. Alsumady, H. Seidel, T. Manzaneque, J. Hernando-García, J.L. Sanchez-Rojas, A. Bittner, U. Schmid, c-axis orientation and piezoelectric coefficients of AlN thin films sputter-deposited on titanium bottom electrodes. Appl. Surf. Sci. 259, 59–65 (2012). https://doi.org/10.1016/j.apsusc.2012.06.086
A. Pandey, R. Prakash, S. Dutta, S. Dalal, A. Kumar, A.K. Kapoor, D. Kaur, Growth and morphological evolution of c-axis oriented AlN films on Si (100) substrates by DC sputtering technique. AIP Conf. Proc. 1953, 1–5 (2018). https://doi.org/10.1063/1.5032964
A. Pandey, S. Dutta, R. Prakash, S. Dalal, R. Raman, A.K. Kapoor, D. Kaur, Growth and evolution of residual stress of AlN films on silicon (100) wafer. Mater. Sci. Semicond. Process. 52, 16–23 (2016). https://doi.org/10.1016/j.mssp.2016.05.004
N. Gupta, A. Pandey, S.R.K. Vanjari, S. Dutta, Influence of residual stress on performance of AlN thin film based piezoelectric MEMS accelerometer structure. Microsyst. Technol. 25, 3959–3967 (2019). https://doi.org/10.1007/s00542-019-04334-1
D. Holec, P.H. Mayrhofer, Surface energies of AlN allotropes from first principles. Scripta Mater. 67, 760–762 (2012). https://doi.org/10.1016/j.scriptamat.2012.07.027
J.X. Zhang, H. Cheng, Y.Z. Chen, A. Uddin, S. Yuan, S.J. Geng, S. Zhang, Growth of AlN films on Si (100) and Si (111) substrates by reactive magnetron sputtering. Surf. Coat. Technol. 198, 68–73 (2005). https://doi.org/10.1016/j.surfcoat.2004.10.075
R. Ruh, A. Zangvil, J. Barlowe, Elastic properties of SiC, AIN, and their solid solutions and particulate composites. Am. Ceram. Soc. Bull. 64, 1368–1373 (1985)
K. Kim, W.R.L. Lambrecht, B. Segall, Elastic constants and related properties of tetrahedrally bonded BN, AlN, GaN, and InN. Phys. Rev. B 53, 16310 (1996). https://doi.org/10.1103/PhysRevB.53.16310
S. Yu, R. Chen, G. Zhang, J. Cheng, Z. Meng, Ferroelectric enhancement in heterostructured ZnO/BiFeO3-PbTiO3 film. Appl. Phys. Lett. 89, 3–6 (2006). https://doi.org/10.1063/1.2393004.1
S. Dutta, A. Pandey, I. Yadav, O.P. Thakur, R. Laishram, R. Pal, R. Chatterjee, Improved electrical properties of PbZrTiO3/BiFeO3 multilayers with ZnO buffer layer. J. Appl. Phys. 112, 084101 (2012). https://doi.org/10.1063/1.4759123
S. Dutta, A. Pandey, O.P. Thakur, R. Pal, R. Chatterjee, Estimation of residual stress in Pb(Zr0.52Ti0.48)O3/BiFeO3 multilayers deposited on silicon. J. Appl. Phys. 114, 174103 (2013). https://doi.org/10.1063/1.4828874
G.G. Stoney, The tension of metallic films deposited by electrolysis. Proc. R. Soc. Lond. A82, 172 (1909). https://doi.org/10.1098/rspa.1909.0021
S. Dutta, A. Pandey, I. Yadav, O.P. Thakur, A. Kumar, R. Pal, R. Chatterjee, Growth and electrical properties of spin coated ultrathin ZrO2 films on silicon. J. Appl. Phys. 114, 014105 (2013). https://doi.org/10.1063/1.4812733
M.J. Madau, Fundamental of Microfabrication-The Science of Miniaturization (CRC Press, Boca Raton, 2002).
A. Pandey, S. Dutta, R. Prakash, R. Raman, A.K. Kapoor, D. Kaur, Growth and comparison of residual stress of AlN films on silicon (100), (110) and (111) substrates. J. Electron. Mater. 47, 1405–1413 (2018). https://doi.org/10.1007/s11664-017-5924-8
A. Pandey, J. Kaushik, S. Dutta, A.K. Kapoor, D. Kaur, Electrical and structural characteristics of sputtered c-oriented AlN thin films on Si (100) and Si (110) substrates. Thin Solid Films 666, 143–149 (2018). https://doi.org/10.1016/j.tsf.2018.09.016
A. Pandey, B.S. Yadav, D.V.S. Rao, D. Kaur, A.K. Kapoor, Dislocation density investigation on MOCVD-grown GaN epitaxial layers using wet and dry defect selective etching. Appl. Phys. A 122, 614 (2016). https://doi.org/10.1007/s00339-016-0143-3
G.N. Sharma, S. Dutta, A. Pandey, S.K. Singh, R. Chatterjee, Influence of nickel doping on structural, morphological and mechanical properties of BiFeO3 thin films. Mater. Chem. Phys. 216, 47–50 (2018). https://doi.org/10.1016/j.matchemphys.2018.05.073
G.N. Sharma, S. Dutta, A. Pandey, S.K. Singh, R. Chatterjee, Microstructure and improved electrical properties of Ti-substituted BiFeO3 thin films. Mater. Res. Bull. 95, 223–228 (2017). https://doi.org/10.1016/j.materresbull.2017.07.046
G. Catalan, J.F. Scott, Physics and applications of bismuth ferrite. Adv. Mater. 21, 2463–2485 (2009). https://doi.org/10.1002/adma.200802849
N.A. Spaldin, S.W. Cheong, R. Ramesh, Multiferroics: past, present, and future. Phys. Today 63, 38 (2010). https://doi.org/10.1557/mrs.2017.86
K. Kaviyarasu, E. Manikandan, J. Kennedy, M. Jayachandran, R. Ladchumananandasiivam, U.U. De-Gomes, M. Maaza, Synthesis and characterization studies of NiO nano-rods for enhancing solar cell efficiency using photon up conversion materials. Ceram. Int. 42, 8385–8394 (2016). https://doi.org/10.1016/j.ceramint.2016.02.054
S. Debnath, P. Predecki, R. Suryanarayanan, Use of glancing angle X-ray powder diffractometry to depth-profile phase transformations during dissolution of in-domethacin and theophylline tablets. Pharm. Res. 21(1), 149–159 (2004). https://doi.org/10.1023/B:PHAM.0000012163.89163.f8
M. Nauer, K. Ernst, W. Kautek, M. Neumann-Spallart, Depth profile characterization of electrodeposited multi-thin-film structures by low angle of incidence X-ray diffractometry. Thin Solid Films 489, 86–93 (2005). https://doi.org/10.1016/j.tsf.2005.05.008
M. Bouroushian, T. Kosanovic, Characterization of thin films by low incidence X-ray diffraction. Cryst. Struct. Theory Appl. 01, 35–39 (2012). https://doi.org/10.4236/csta.2012.13007
C. Kumari, A. Pandey, A. Dixit, Zn interstitial defects and their contribution as efficient light blue emitters in Zn rich ZnO thin films. J. Alloy Compd. 735, 2318–2323 (2018). https://doi.org/10.1016/j.jallcom.2017.11.377
M. Agrawal, A. Jain, D.V. SridharaRao, A. Pandey, A. Goyal, A. Kumar, S. Lamba, B.R. Mehta, K. Muraleedharan, R. Muralidharan, Nanoharvesting of GaN nanowires on Si (211) substrates by plasma-assisted molecular beam epitaxy. J. Cryst. Growth. 402, 37–41 (2014). https://doi.org/10.1016/j.jcrysgro.2014.05.004
S.K. Jangir, H.K. Malik, S. Dalal, A. Pandey, T. Srinivasan, K. Muraleedharan, R. Muralidharan, P. Mishra, X-ray pole figure analysis of catalyst free InAs nanowires on Si substrate. Mater. Sci. Eng. B 225, 108–114 (2017). https://doi.org/10.1016/j.mseb.2017.08.017
J. IlHong, J. Bae, Z.L. Wang, R.L. Snyder, Room-temperature, texture-controlled growth of ZnO thin films and their application for growing aligned ZnO nanowire arrays. Nanotechnology. 20, 085609 (2009). https://doi.org/10.1088/0957-4484/20/8/085609
S.K. Jain, R.R. Kumar, N. Aggarwal, P. Vashishtha, L. Goswami, S. Kuriakose, A. Pandey, M. Bhaskaran, S. Walia, G. Gupta, Current transport and band alignment study of MoS2/GaN and MoS2/AlGaN heterointerfaces for broadband photodetection application. ACS Appl. Electron. Mater. 2, 710–718 (2020). https://doi.org/10.1021/acsaelm.9b00793
A. Pandey, S. Dutta, A. Kumar, R. Raman, A.K. Kapoor, R. Muralidhran, Structural and optical properties of bulk MoS2 for 2D layer growth. Adv. Mater. Lett. 7, 777–782 (2016). https://doi.org/10.5185/amlett.2016.6364
Y. Huang, G. Pandraud, P.M. Sarro, Characterization of low temperature deposited atomic layer deposition TiO2 for MEMS applications. J. Vac. Sci. Technol. A. 31, 01A148 (2013). https://doi.org/10.1116/1.4772664
B. Wang, J. Yan, H. Cui, S. Du, Preparation and characterization of nano TiO2/micron Cr2O3 composite particles, J. Alloys Compd. 509, 5017–5019 (2011). https://doi.org/10.1016/j.jallcom.2011.02.008
T. Jantson, T. Avarmaa, H. Mandar, T. Uustare, R. Jaaniso, Nanocrystalline Cr2O3–TiO2 thin films by pulsed laser deposition. Sens. Actuators B 109, 24–31 (2005). https://doi.org/10.1016/j.snb.2005.03.014
S. Dutta, A. Pandey, Leeladhar, K.K. Jain, Growth and characterization of ultrathin TiO2–Cr2O3 nano-composite films. J. Alloy Compd. 696, 376–381 (2017). https://doi.org/10.1016/j.jallcom.2016.11.284
B. Mehdikhani, G.H. Borhani, S.R. Bakhshi, H.R. Baharvandi, Synthesis of tantalum carbide/boride nanocomposite powders by mechanochemical method. Int. J. Eng. Trans. B Appl. 27, 769–774 (2014). https://doi.org/10.5829/idosi.ije.2014.27.05b.13
N. Khemiri, D. Abdelkader, B. Khalfallah, M. Kanzari, Synthesis and characterization of CuIn2n+1S3n+2 (with n = 0, 1, 2, 3 and 5) powders. J. Synth. Theory Appl. 02, 33–37 (2013). https://doi.org/10.4236/ojsta.2013.21003
J. Wieben, C. Beckmann, H. Yacoub, A. Vescan, H. Kalisch, Development of a III-nitride electro-optical modulator for UV–Vis. Jpn. J. Appl. Phys. 58, 7–12 (2019). https://doi.org/10.7567/1347-4065/ab079e
J. Shan, S.L. Dexheimer, Time-resolved terahertz studies of conductivity processes in novel electronic materials. Terahertz Spectrosc. Princ. Appl. (2017). https://doi.org/10.1201/9781420007701
Acknowledgements
The authors acknowledge Dr. Seema Vinayak, Director, SSPL, for her continuous support and for the permission to publish this review article. Help from other colleagues of SSPL are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pandey, A., Dalal, S., Dutta, S. et al. Structural characterization of polycrystalline thin films by X-ray diffraction techniques. J Mater Sci: Mater Electron 32, 1341–1368 (2021). https://doi.org/10.1007/s10854-020-04998-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-04998-w