
Abstract
The specific surface area is an important parameter influencing the storage capability of materials due to its direct effect on the reaction sites availability. In this study, ZnFe2O4 nanoparticles with a high specific surface area, 78.9 m2 g−1, were prepared by a weak ultrasonic irradiation technique. Fe(NO3)3·9H2O, Zn(NO3)2·6H2O and glucose were used as reagents and the prepared precursor was calcined in the air at 400 °C for 3 h. The product was characterized by thermogravimetric analysis, Fourier transform infrared, Raman spectra, X-ray diffraction, Brunauer–Emmett–Teller, Scanning electron microscopy as well as Transmission electron microscopy. The charge storage ability, cycling stability, and ion transport of the produced ZnFe2O4 nanoparticles were examined by cyclic voltammetry, galvanostatic charge–discharge and electrochemical impedance spectroscopy tests in 1, 3 and 6 M KOH solutions. The highest specific capacitance of the ZnFe2O4 nanoparticles was obtained 712 F g−1 at the scan rate of 2 mV s−1 in 6 M KOH electrolyte, and the capacity retention for ZnFe2O4 nanoparticles was still maintained after 2000 cycles at 92.4, 94.2 and 96.6% in 1, 3 and 6 M KOH electrolytes, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Increasing demand for nanomaterials with large specific surface area for a variety of uses (e.g., supercapacitors) has recently attracted the attention of researchers in this field [1]. Supercapacitors are tools with high power density, long cycle life and high charging and discharging rates [2,3,4,5,6,7,8,9,10,11]. There are growing demands for the next-generation flexible and lightweight energy storage devices with high power and energy densities due to their unique characteristics and potential applications in various portable electronic tools such as memory backup systems [3] smart sensors, flexible touch screen, electronic newspaper [5, 6], mobiles [5, 6, 9] and hybrid electric vehicles [3, 8, 9]. Supercapacitors can be classified as electrical double-layer capacitors (EDLCs) according to the energy storage mechanism, which store energy through the formation of electrical double layer at the electrode–electrolyte interface (e.g., carbon-based materials) and pseudocapacitor, which store energy by electron transfer Faradaic redox reaction [7, 8, 12,13,14,15,16,17,18]. Transition metal oxides having binary and ternary compositions like RuO2, NiO, CuO, CoO, Co3O4, MnO2, Mn3O4 [16, 19], NiMoO4, Fe3O4, Fe2O3, MnFe2O4, ZnFe2O4, CuFe2O4, CoFe2O4 NiCo2O4, and CuCo2O4 have been widely studied as the pseudocapacitor electrode materials [16]. The synthesis method of electrode materials are key challenges because their low energy density compared to batteries prevents their use as a major power source. High energy and power along with better structural stability are important parameters that must be considered before synthesizing a supercapacitor electrode material [20]. To improve the energy density and overall performance, mixed transition metal oxides (MTMO) with two different metals have been studied widely as supercapacitors. The chemical composition and the synergic effect of multiple oxidation states of the metal cations in MTMOs are important to get the satisfied electrochemical behavior [5, 6]. Among these MTMOs, ZnFe2O4 is the most promising due to its abundancy [4], low toxicity [4,5,6], low-cost [4, 5, 16], environmental benignity behavior [5, 6], excellent chemical, thermal stability and low resistance [5]. There are various ways to prepare ZnFe2O4 nanostructures, such as co-precipitation [19, 21,22,23], ball milling [19, 22, 23], sol–gel [19, 21, 23], hydrothermal [19, 21,22,23], thermal decomposition [19], ceramic method [19, 24], electrodeposition [21], ultrasonic [22] and combustion method [22, 24]. Few studies have used sonochemical method for the ZnFe2O4 preparation. In this study, for supercapacitor application, ZnFe2O4 nanoparticles with high specific surface area were synthesized in ultrasonic bath. Among alternative methods of controlling synthesis, the sonochemical method was found to produce nanomaterials with smaller size and larger surface area than those reported in previous studies [25, 26]. In fact, this method is an effective, useful, low-cost and simple technique.
2 Experimental
2.1 Materials
All chemicals were purchased from Merck Company (China) and used with no further purification. In all the experiments, double-distilled water was used.
2.2 Preparation of ZnFe 2 O 4
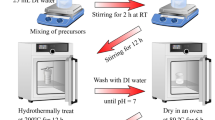
In the typical synthesis of ZnFe2O4, stoichiometric amounts of Zn(NO3)2·6H2O (3 mmol) and Fe(NO3)3·9H2O (6 mmol) were dissolved separately in (20 mL) doubly distilled water to form a clear solution and then mixed together, followed by the adding 1 g glucose and 2 mL mono ethanol amine under stirring. The pH value of the obtained solution was set to 10 ± 0.5 using ammonia. Then, this solution was subjected to low ultrasound irradiation for 30 min under continuous stirring in the ultrasonic bath. Once the reaction was finished and naturally cooled to room temperature, the dark precipitate was collected by centrifugation and washed with water for several times. Then, it was dried in an oven at 60 °C and calcined for 3 h at 400 °C.
2.3 Characterization
Using thermogravimetric analysis (TGA), the mass changes of precursor was evaluated at a temperature of 25–900 °C and a heating rate of 5 °C per minute under air atmosphere using NETZSCH TG 209 F1 Iris device. The X-ray diffraction (XRD) analysis was used to study the crystalline structure of the samples using a STOE XD-3A diffractometer using CuKα radiation at λ = 1.5406 Å. For characterizing morphologies, a Zeiss scanning electron microscope and a Zeiss-EM10C transmission electron microscope were used. The Fourier transform infrared (FT-IR) spectra of samples were recorded using a SHIMADZU-8400S-series FT-IR spectrometer on KBr disks within the range of 400–4000 cm−1. Raman spectroscopy was performed at room temperature using a charge-coupled device spectrometer for signal detection and a Nd:YAG crystal laser (power = 10–90 mW). The BET surface area and pore size distribution of the samples were assessed through nitrogen adsorption/desorption isotherms measured at − 196 °C using a Micromeritics ASAP 2020 system.
2.4 Electrochemical measurements
An autolab potentiostat/galvanostat (PGSTAT 204) was used for the electrochemical measurements including cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) tests. These measurements were performed using a standard three-electrode configuration, where a platinum wire was used as counter electrode, an Ag/AgCl reference electrode, and a Ni foam (with 1 cm2 area) working electrode. The ZnFe2O4 nanoparticles’ capacitive behaviors were characterized by CV in 1, 3, and 6 M KOH electrolyte within the potential range from − 0 to 0.5 V at room temperature compared to Ag/AgCl at different scanning rates of 1–100 mV s−1. Then, the specific capacitance (SC) values were determined by integrating the recorded current over the applied potential range according to the following equation:
where I = the measured current (A); Va–Vc = the potential sweep range (V); m = the mass of electrochemically active material (g); and v = the sweep rate (V S−1).
3 Results and discussion
3.1 TGA and XRD analysis
To obtain calcination temperature, the TGA was performed under air atmosphere at the temperature range 25–900 °C and an heating rate of 5 °C per minute (Fig. 1a). The mass changes were observed in three steps. Based on this curve, the first step, the weight loss of 2.42% occurs at 100 ºC due to the release of adsorbed water molecules. The second and third stages of 41.19% weight loss at 100–380 ºC can be due to lattice water molecule evaporation and organic matter decomposition. Almost no weight loss could be observed during 380–900 °C, indicating the precursor’s complete decomposition at temperatures above 400 °C.

TGA analysis (a) and XRD pattern of ZnFe2O4 nanoparticles (b)
Structural analysis of ZnFe2O4 nanostructures was carried out by XRD. The XRD pattern of ZnFe2O was given in Fig. 1b. According to this figure, evolved diffraction peaks could be indexed to a cubic phase of ZnFe2O4 (S.G.: Fd-3m) and the lattice parameters were obtained such as a, b and c = 8.4409 Å (JCPDS card No. 01-082-1049). The appeared diffraction peaks at angles 18.189, 29.917, 35.236, 42.819, 53.112, 56.613,62.160, 70.503, 73.513, and 74.506° matched with the (111), (220), (311), (400), (422), (333), (440), (620), (533), and (622) crystal planes of cubic ZnFe2O4, which confirms the formation of ZnFe2O4 compound with excellent crystallinity with no other impurity peaks. The lack of any more by product shows the sample purity.
3.2 FT-IR and Raman analysis
Figure 2a shows the FT-IR spectrum of ZnFe2O4 nanoparticles recorded over the wavenumber range from 4000 to 400 cm−1 at room temperature. The observed broad band at 3323 cm−1 belongs to the O–H stretching vibrations of the adsorbed water molecules [22, 27]. The appeared peaks at 438 and 553 cm−1 are corresponded to the intrinsic lattice vibrations of Fe–O and Zn–O, octahedral and tetrahedral coordination compounds in the spinel structure of ZnFe2O4, respectively [22, 28]. Figure 2b presents Raman spectroscopy results for the ZnFe2O4 nanoparticles in the area from 200 to 800 cm−1 at room temperature. The normal spinel structure (AB2O4) had 5 active Raman peaks at 271, 350, 505, 647 and 693 cm−1 related to the Raman modes (A1g + Eg + 3T2g) in ZnFe2O4 cubic spinel. The low-frequency mode at 271 cm−1, which represents the characteristics of octahedral sites (BO6), is assigned to Eg symmetry, and the appeared modes at 350 and 476 cm−1 are assigned to T2g symmetry. The high-frequency modes at 647 and 693 cm−1 are related to the A1g symmetry, which includes the O atom vibrations in AO4 tetrahedral groups. Hence, the observed frequency mode at 647 cm−1 is related to the oxygen vibrations against zinc, while the other frequency mode at 693 cm−1 is because of the corresponding oxygen vibrations against iron [29].

FT-IR spectrum (a) and Raman spectrum of the ZnFe2O4 nanoparticles (b)
3.3 Morphology
The morphology and the size of product were investigated by means of SEM and TEM. The SEM image of ZnFe2O4 nanoparticles has given in Fig. 3a. The TEM image (Fig. 3b) well showed the nanometer sized ZnFe2O4 particles, which strongly support SEM analysis. The histogram of the particle size distribution based on SEM images is illustrated in Fig. 3c. The diameter of most nanoparticles ranged from 30 to 40 nm. The results of energy dispersive X-ray spectroscopy (EDX) analysis (Fig. 3d) confirm the presence of only iron, zinc, and oxygen elements in the nanoparticles.

SEM images (a) TEM image (b) Size distribution histogram (c) and the energy dispersive EDX of ZnFe2O4 nanoparticles (d)
3.4 BET surface area analysis
The specific surface area is an essential parameter influencing the storage capability of electrode materials due to its direct effect on the reaction sites availability [30]. The specific surface area and pore size of ZnFe2O4 nanoparticles were estimated using the BET and BJH methods, respectively. Pore structure of all samples were analyzed by N2 adsorption at − 196 °C. Before each measurement, sample was evacuated 3 h at 300 °C. The nitrogen adsorption/desorption isotherms of ZnFe2O4 nanoparticles and corresponding pore size distribution curves have been shown in Fig. 4a, b. In Fig. 4a, nanoparticles had type IV isotherm with hysteresis loops in a relative pressure (P/P0) range of 0.6–0.9, indicating the mesoporous structure formation [31]. The observed BET surface area of the ZnFe2O4 nanoparticles was 78.9 m2 g−1. High surface area in the ZnFe2O4 nanoparticles led to an enhanced electrochemical capacity of these nanoparticles. The Barrett–Joyner–Halenda (BJH) analysis results showed a narrow pore size distribution centered at 8 nm (Fig. 4b). The average pore diameters of the ZnFe2O4 nanoparticles were reported to be in the mesoporous area, which increases the diffusion of ions to ZnFe2O4 samples, leading to a higher electrochemical performance [30].

Nitrogen adsorption/desorption isotherm (a) and corresponding pore size distribution plots of ZnFe2O4 nanostructures (b)
3.5 Electrochemical properties
As mentioned before, the study of the electrochemical performance of the synthesized sample, as electroactive electrode, was examined by CV, GCD and EIS methods in KOH aqueous solution (1 M, 3 M and 6 M) under a standard three-electrode system. To provide working electrode, 80 wt.% of the prepared sample (0.01 g) was mixed with 15 wt% acetylene black as a conducting material and 5 wt% of polyvinylidene fluoride as binder in N-methyl-2-pyrrolidone solvent. The obtained paste was then loaded on the Ni foam electrode surface and dried at 70°C for 12 h. To investigate the effect of electrolyte concentration on the superconducting characteristics of the synthesized sample, electrolyte selection was performed according to Yang et al. [6].
Figure 5a shows the CV curves of the ZnFe2O4 nanoparticles electrode in the potential window of 0–0.5 V vs. Ag/AgCl at scan rate of 50 mV s−1 in 1, 3 and 6 M KOH aqueous electrolytes. Also, show the CV curve of the blank in 6 M KOH aqueous electrolyte and other conditions mentioned. The anodic and cathodic peaks emerged in the CV curve because of the oxidation and reduction process of Zn and Fe ions accompanied with OH¯ ion of the electrolyte. The possible redox reaction takes place in the electrode as follows [32]:

CV curves of ZnFe2O4 nanoparticles nanostructures at scan rate of 50 mVs−1 in 1, 3 and 6 M KOH aqueous electrolytes (a), CV curves of ZnFe2O4 nanoparticles nanostructures at a scan rate of 5 to 100 mV s−1 in 6 M KOH aqueous electrolyte. (b), the calculated SC for ZnFe2O4 nanoparticles electrode at the potential sweep rate from 2 to 200 mV s−1
Based on Eq. (1), the SC values at 50 mV s−1 were obtained 187, 301 and 455 F g−1 in 1, 3 and 6 M KOH aqueous electrolytes, respectively. A higher SC was observed with the increase in electrolyte concentration. The CV curves of the ZnFe2O4 electrode in 1, 3 and 6 M KOH electrolytes were symmetrical according to the zero current line and a rapid current change around the potential reversal at the end each scan, which shows the ideal pseudocapacitive behavior of the study sample. The SC value of the blank was obtained 63 F g−1 at the scan rate of 50 mV s−1 in 6 M KOH electrolyte. Figure 5b shows the CV curves of the ZnFe2O4 nanoparticles electrode at a scan rate of 5, 10, 20, 30, 50, 75 and 100 mV s−1 in 6 M KOH electrolyte. According to Fig. 5c, the SC for the ZnFe2O4 nanoparticles was decreased from 712 to 348, 516 to 212 and 378.3 to 131 F g−1 in 6, 3 and 1 M KOH aqueous electrolytes with a potential sweep rate of 2–200 mV s−1, respectively. It can be assumed that the electrolyte ions of K+ or OH− had enough time to move through the available pores on the selected surface at low scan rates, resulting in a more effective redox reaction or a higher SC value. However, when the scan rate was increased up to 200 mV s−1, their time became limited and thus the interaction between the electrolyte ions and the pores was significantly declined. High values obtained for SC can be due to the significant increase in the specific surface area in the synthesized sample [6]. Based on the CV values, supercapacitive performance of ZnFe2O4 electrode in 6 M KOH electrolyte was better compared to others presented in Table 1. Also, the results obtained are comparable to the results obtained from other electrode materials with high SC [36,37,38,39,40].
Cycling stability of the electrode is one of the most important factors for practical use of supercapacitors. For ZnFe2O4 electrode, its cyclic stability was tested for 2000 cycles in 1, 3 and 6 M KOH electrolytes at a scan rate of 50 mV s−1. The results showed that all electrodes had very high cyclic stability (Fig. 6). The SC values were decreased by 7.6%, 5.8% and 3.4% in 1 M, 3 M and 6 M KOH electrolytes after applying 2000 CVs, respectively. It indicates the high stability of ZnFe2O4 electrode in 6 M KOH electrolyte, proving that it is suitable for application in fast-charging devices.

Cycling stability of ZnFe2O4 nanoparticles obtained from CV measurements at the scan rate of 50 mV s−1 at 1 M KOH (a) 3 M KOH (b) and 6 M KOH (c)
To detect possible changes in the structure of ZnFe2O4 nanoparticles after stability test, the XRD patterns of the Ni foam and electrode were recorded before and after stability test (Fig. 7).

XRD patterns of the Ni foam (a), the electrode made of ZnFe2O4 nanoparticles before (b) and after (c) the stability test
Figure 7a–c represents the XRD patterns of the Ni foam, the electrode made of ZnFe2O4 nanoparticles before and after the stability test, respectively. According to this figure, the XRD patterns of the electrode, before and after the stability test, show that the structure of ZnFe2O4 nanoparticles has not changed after stability test. However, in the XRD pattern of the electrode, after the stability test, the intensity of the material peaks has been reduced. Figure 8a and b shows the SEM images of the electrode made of ZnFe2O4 nanoparticles before the stability test and the same electrode after the stability test, respectively. It seems that after the stability test, the morphology of some of the material has changed and the nanoparticles have turned into nanoflowers. Nanoflowers are larger than nanoparticles and probably have a smaller specific surface area. Morphological changes can reduce the specific capacitance [30, 41].

SEM images of the electrode made of ZnFe2O4 nanoparticles before the stability test (a) and the same electrode after the stability test (b)
Galvanostatic charge–discharge performance was tested to evaluate the supercapacitive properties of ZnFe2O4 nanoparticles as electrode materials. Figure 9a shows the charge–discharge curves of the ZnFe2O4 electrode in 1, 3 and 6 M KOH electrolytes at 2 A g−1 current density and within the potential range of 0–0.5 V vs. Ag/AgCl. Also, show the charge–discharge curve of the nickel foam in 6 M KOH aqueous electrolyte and other conditions mentioned. The SC was defined using the following equation:

GCD curves of ZnFe2O4 nanoparticles electrode in 1 M, 3 M and 6 M KOH electrolytes at the current density of 2 A g−1 (a), GCD curves of ZnFe2O4 nanoparticles electrode at current densities of 1, 2, 4, 8 and 16 A g−1 in 6 M KOH aqueous electrolyte (b), the calculated SC for ZnFe2O4 nanoparticles electrode, at current densities from 1 to 32 A g−1 (c) Ragone plots obtained for ZnFe2O4 nanoparticles electrode in 1, 3 and 6 M KOH (d)
where I = the discharge current (A), ΔV = the potential window (V), Δt = the discharge time (s) and m = the mass of electroactive material (g).
The SC for our sample at 1 A g−1 current density and 1, 3 and 6 M KOH electrolytes were calculated 364, 496 and 680 F g−1, respectively. As can be seen, the values are in agreement with the SC values calculated based on recorded CV, and the time of charge–discharge increases with the increase in electrolyte concentration. The SC value of the blank was obtained 104 F g−1 at 2 A g−1 current density and 6 M KOH electrolyte. Figure 9b shows the charge–discharge curves for characterizing the capacitive performance of the ZnFe2O4 electrode in the potential window of 0–0.5 V vs. Ag/AgCl at current densities of 1, 2, 4, 8 and 16 A g−1 in 6 M KOH aqueous electrolyte. As can be seen, with the increase of current density, the charge–discharge time becomes shorter. The SC values for ZnFe2O4 electrode at current densities from 1 to 32 A g−1 are presented in Fig. 9c. The SC value was decreased from 696 to 320, from 506 to 256, and from 372 to 179 F g−1 in 6, 3 and 1 M KOH aqueous electrolytes, respectively. Figure 9d presents the Ragone plot of power density versus energy density, obtained from the charge–discharge analysis at different current densities. The maximum energy density (24.2 W h kg−1) was obtained in tests using 6 M KOH aqueous electrolyte at 250 W kg−1 power density.
To evaluate the ion transport mechanism, EIS test was performed in the frequency range of 105–0.01 Hz at open circuit potential. Figure 10 illustrates the Nyquist plots and the equivalent circuit (inset) for ZnFe2O4 nanoparticles, showing the ion diffusion and electron transfer in 1, 3 and 6 M KOH solutions. At the high-frequency intercept, the real axis value is represented by Rs, including the electrolyte’s ionic resistance, the active materials’ intrinsic resistance, and the contact resistance at the interface of electrodes and current collector [30]. As shown in Fig. 10, the ZnFe2O4 electrode spectra consists of two parts; one charge transfer resistance (Rct)-related semicircle at the high-frequency region related to the redox reactions and one straight line in the low-frequency region represented by Warburg impedance (W), which is related to the electrolyte diffusion within the ZnFe2O4 nanoparticles. Cdl represents the electrical double-layer capacitance at electrode–electrolyte interface, while CF shows the pseudocapacitance resulted from the faradic reactions [30, 42]. The values obtained for each equivalent circuit are given in Table 2. As can be seen, the Rct value in the 6 M KOH electrolyte is less than those in the 3 and 1 M electrolytes, indicating that the process of charge transfer improves by increasing the electrolyte concentration. The Rs value was almost constant for all of the tested electrolytes. According to CV test and charge–discharge results, it is expected that the CF value for the test in 6 M KOH electrolyte be higher than that in other electrolytes. For this reason, the test in 6 M KOH solution showed better electrochemical performance than in other electrolytes (Table 2). In fact, the redox reaction was better in the electrolyte with a higher concentration.

Nyquist plots of ZnFe2O4 nanoparticles in 1, 3 and 6 M KOH solution; the inset shows the expanded high-frequency region
4 Conclusion
In the present study, ZnFe2O4 nanoparticles with high specific surface area were prepared using a weak ultrasonic irradiation technique. The diameter of most ZnFe2O4 nanoparticles was in a range from 30 to 40 nm. The study of electrochemical performance of the synthesized sample, as electroactive electrode, was performed by CV, GCD and EIS in KOH aqueous solution (1 M, 3 M and 6 M). The highest SC for ZnFe2O4 nanoparticles was 712 F g−1 in 6 M KOH aqueous electrolyte at scan rate of 2 mV s−1, which is better than the value reported in studies. Based on the results, it is suggested that the ZnFe2O4 nanoparticles be used as a promising material to improve the performance of SC electrodes.
References
S. Dhanalakshmi, A. Mathi Vathani, V. Muthuraj, N. Prithivikumaran, S. Karuthapandian, Mesoporous Gd2O3/NiS2 microspheres: a novel electrode for energy storage applications. J. Mater. Sci. Mater. Electron. (2020). https://doi.org/10.1007/s10854-020-02858-1
K. Mohamed Racik, K. Guruprasad, M. Mahendiran, J. Madhavan, T. Maiyalagan, M. Victor Antony Raj, Enhanced electrochemical performance of MnO2/NiO nanocomposite for supercapacitor electrode with excellent cycling stability. J. Mater. Sci. Mater. Electron. 30, 5222–5232 (2019). https://doi.org/10.1007/s10854-019-00821-3
B.G.S. Raj, A.M. Asiri, A.H. Qusti, J.J. Wu, S. Anandan, Sonochemically synthesized MnO2 nanoparticles as electrode material for supercapacitors. Ultrason Sonochem 21, 1933–1938 (2014). https://doi.org/10.1016/j.ultsonch.2013.11.018
F.O. Agyemang, H. Kim, Electrospun ZnFe2O4-based nanofiber composites with enhanced supercapacitive properties. Mater Sci Eng B 211, 141–148 (2016). https://doi.org/10.1016/j.mseb.2016.06.011
S.S. Raut, B.R. Sankapal, First report on synthesis of ZnFe2O4 thin film using successive ionic layer adsorption and reaction: approach towards solid-state symmetric supercapacitor device. Electrochim. Acta 198, 203–211 (2016). https://doi.org/10.1016/j.electacta.2016.03.059
Y. Lin, J. Wang, H. Yang, L. Wang, M. Cao, Comparative study of individual aqueous electrolytes with porous ZnFe2O4 for supercapacitor. Mater. Lett. 207, 195–197 (2017). https://doi.org/10.1016/j.matlet.2017.07.075
S.M.B. Malek Hosseini, S.M. Baizaee, H.R. Naderi, A.D. Kordi, Excimer laser assisted very fast exfoliation and reduction of graphite oxide at room temperature under air ambient for supercapacitors electrode. Appl. Surf. Sci. 427, 507–516 (2018). https://doi.org/10.1016/j.apsusc.2017.08.029
G.K. Veerasubramani, K. Krishnamoorthy, R. Sivaprakasam, S.J. Kim, Sonochemical synthesis, characterization, and electrochemical properties of MnMoO4 nanorods for supercapacitor applications. Mater. Chem. Phys 147, 836–842 (2014). https://doi.org/10.1016/j.matchemphys.2014.06.028
S.E.M. Pourhosseini, O. Norouzi, P. Salimi, H.R. Naderi, Synthesis of a novel interconnected 3D pore network algal biochar constituting iron nano particles derived from a harmful marine biomass as high performance asymmetric supercapacitor electrodes. ACS Sustain. Chem. Eng. 6, 4746–5475 (2018). https://doi.org/10.1021/acssuschemeng.7b038718
K. Tul Kubra, R. Sharif, B. Patil, A. Javaid, S. Shahzadi, A. Salman, S. Siddique, G. Ali, Hydrothermal synthesis of neodymium oxide nanoparticles and its nanocomposites with manganese oxide as electrode materials for supercapacitor application. J. Alloys Compd. 815, 152104 (2020). https://doi.org/10.1016/j.jallcom.2019.152104
B. Kirubasankar, V. Murugadoss, S. Angaiah, Hydrothermal assisted in situ growth of CoSe onto graphene nanosheets as a nanohybrid positive electrode for asymmetric supercapacitors. RSC Adv. 7, 5853–5862 (2017). https://doi.org/10.1039/c6ra25078e
S. Arunachalam, B. Kirubasankar, E. Rajagounder Nagarajan, D. Vellasamy, S. Angaiah, A facile chemical precipitation method for the synthesis of Nd(OH)3 and La(OH)3 nanopowders and their supercapacitor performances. ChemistrySelect 3, 12719–12724 (2018). https://doi.org/10.1002/slct.201803151
K. Balakrishnan, M. Kumar, A. Subramania, Synthesis of polythiophene and its carbonaceous nanofibers as electrode materials for asymmetric supercapacitors. Adv. Mater. Res 938, 151–157 (2014). https://doi.org/10.4028/www.scientific.net/AMR.938.151
B. Kirubasankar, V. Murugadoss, J. Lin, T. Ding, M. Dong, H. Liu, J. Zhang, T. Li, N. Wang, Z. Guo, S. Angaiah, In situ grown nickel selenide on graphene nanohybrid electrodes for high energy density asymmetric supercapacitors. Nanoscale 10(2018), 20414–20425 (2018). https://doi.org/10.1039/c8nr06345a
J. Yesuraj, V. Elumalai V, Bhagavathiachari M, Samuel AS, Elaiyappillai E, Johnson PM, , a facile sonochemical assisted synthesis of α-MnMoO4/PANI nanocomposite electrode for supercapacitor applications. J. Electroanal. Chem. 797, 78–88 (2017). https://doi.org/10.1016/j.jelechem.2017.05.019
M.M. Vadiyar, S.S. Kolekar, J.Y. Chang, A.A. Kashale, A.V. Ghule, Reflux condensation mediated deposition of Co3O4 nanosheets and ZnFe2O4 nanoflakes electrodes for flexible asymmetric supercapacitor. Electrochim. Acta 222, 1604–1615 (2016). https://doi.org/10.1016/j.electacta.2016.11.146
H. Xiao, S. Yao, H. Liu, F. Qu, X. Zhang, X. Wu, NiO nanosheet assembles for supercapacitor electrode materials. Prog. Nat. Sci. 26, 271–275 (2016). https://doi.org/10.1016/j.pnsc.2016.05.007
D. Lee, Q.X. Xia, R.S. Mane, J.M. Yun, K.H. Kim, Direct successive ionic layer adsorption and reaction (SILAR) synthesis of nickel and cobalt hydroxide composites for supercapacitor applications. J. Alloys Compd. 722, 809–817 (2017). https://doi.org/10.1016/j.jallcom.2017.06.170
S. Ozcan, B. Kaynar, M.M. Can, T. Fırat, Synthesis of ZnFe2O4 from metallic zinc and iron by wet-milling process. Mater. Sci. Eng. B 121, 278–281 (2005). https://doi.org/10.1016/j.mseb.2005.04.006
K. Tul Kubra, A. Javaid, R. Sharif, G. Ali, F. Iqbal, A. Salman, F. Shaheen, A. Butt, F. Jan Iftikhar, Facile synthesis and electrochemical study of a ternary hybrid PANI/ GNP/MnO2 as supercapacitor electrode material. J. Mater. Sci. Mater. Electron. (2020). https://doi.org/10.1007/s10854-020-03792-y
J. Sui, C. Zhang, J. Li, W. Cai, Facile synthesis of multifunctional ZnFe2O4 nanoparticles in liquid polyols. J. Nanosci. Nanotechnol. 12, 3867–3872 (2012). https://doi.org/10.1166/jnn.2012.5872
A. Shanmugavani, R. KalaiSelvan, S. Layek, C. Sanjeeviraja, Size dependent electrical and magnetic properties of ZnFe2O4 nanoparticles synthesized by the combustion method: comparison between aspartic acid and glycine as fuels. J. Magn. Magn. Mater. 354, 363–371 (2014). https://doi.org/10.1016/j.jmmm.2013.11.018
H. Yang, X. Zhang, C. Huang, W. Yang, G. Qiu, Synthesis of ZnFe2O4 nanocrystallites by mechanochemical reaction. J. Phys. Chem. Solids 65, 1329–1332 (2004). https://doi.org/10.1016/j.jpcs.2004.03.001
C.A. Ladole, Preparation and characterization of spinel zinc ferrite ZnFe2O4. Int. J. Chem. Sci 10, 1230–1234 (2012)
S. Anandan, B.G.S. Raj, G.J. Lee, J.J. Wu, Sonochemical synthesis of manganese (II) hydroxide for supercapacitor applications. Mater. Res. Bull. 48, 3357–3361 (2013). https://doi.org/10.1016/j.materresbull.2013.05.021
A. Gedanken, Using sonochemistry for the fabrication of nanomaterials. Ultrason. Sonochem. 11, 47–55 (2004). https://doi.org/10.1016/j.ultsonch.2004.01.037
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th edn. (Wiley, Hoboken, NJ, 2009).
P. Suppuraj, G. Thirunarayanan, M. Swaminathan, I. Muthuvel, Facile synthesis of spinel nanocrystalline ZnFe2O4: enhanced photocatalytic and microbial applications. Mater. Sci. Appl. Chem. 34, 5–11 (2017). https://doi.org/10.1515/msac-2017-0001
M. Maletin, E.G. Moshopoulou, A.G. Kontos, E. Devlin, A. Delimitis, V.T. Zaspalis, L. Nalbandian, V.V. Srdic, Synthesis and structural characterization of In-doped ZnFe2O4 nanoparticles. J. Eur. Ceram. Soc. 27, 4391–4394 (2007). https://doi.org/10.1016/j.jeurceramsoc.2007.02.165
B. Ameri, S.S.H. Davarani, R. Roshani, H.R. Moazami, A. Tadjarodi, A flexible mechanochemical route for the synthesis of copper oxide nanorods/nanoparticles/nanowires for supercapacitor applications: the effect of morphology on the charge storage ability. J. Alloys Compd. 695, 114–123 (2017). https://doi.org/10.1016/j.jallcom.2016.10.144
K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquerol, T. Siemieniewska, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 57, 603–619 (1985). https://doi.org/10.1351/pac198254112201
B. Palanivel, S.D. Mudisoodum Perumal, T. Maiyalagan, V. Jayarman, C. Ayyappan, M. Alagiri, Rational design of ZnFe2O4/g-C3N4 nanocomposite for enhanced Photo-Fenton reaction and supercapacitor performance. Appl. Surf. Sci. 498, 143807 (2019). https://doi.org/10.1016/j.apsusc.2019.143807
Y. Lin, J. Zhang, M. Li, L. Wang, H. Yang, An excellent strategy for synthesis of coral-like ZnFe2O4 particles for capacitive pseudocapacitors. J. Alloys Compd. (2017). https://doi.org/10.1016/j.jallcom.2017.07.328
S. Yang, Z. Han, F. Zheng, J. Sun, Z. Qiao, X. Yang, L. Li, C. Li, X. Song, B. Cao, ZnFe2O4 nanoparticles-cotton derived hierarchical porous active carbon fibers for high rate-capability supercapacitor electrodes. Carbon (2018). https://doi.org/10.1016/j.carbon.2018.03.071
D. Zhang, W. Li, R. Ye, X. Guo, S. Wang, X. Wang, Q. Xiang, A facile strategy for ZnFe2O4 coating preparing by electrophoretic deposition and its supercapacitor performances. J. Mater. Sci. Mater. Electron. 29, 5454–5458 (2018). https://doi.org/10.1007/s10854-017-8512-z
D.K. Maurya, B. Balan, V. Murugadoss, C. Yan, S. Angaiah, A fast Li-ion conducting Li7.1La3Sr0.05Zr1.95O12 embedded electrospun PVDF-HFP nanohybrid membrane electrolyte for all-solid state Li-ion capacitors. Mater. Today Commun. (2020). https://doi.org/10.1016/j.mtcomm.2020.101497
A. Subasri, B. Kirubasankar, V. Vignesh Murugadoss, V. Devadoss, S. Angaiah, Facile synthesis of electrostatically anchored Nd(OH)3 nanorods onto graphene nanosheets as high capacitance electrode material for supercapacitors. New J. Chem. 42, 2923–2932 (2018). https://doi.org/10.1039/C7NJ04335J
B. Kirubasankar, S. Vijayan, S. Angaiah, Sonochemical synthesis of a 2D 2D MoSe2/graphene nanohybrid electrode material for asymmetric supercapacitors. Sustain. Energy Fuels 3, 467–477 (2019). https://doi.org/10.1039/c8se00446c
B. Kirubasankar, P. Palanisamy, S. Arunachalam, V. Murugadoss, S. Angaiah, 2D MoSe2-Ni(OH)2 nanohybrid as an efficient electrode material with high rate capability for asymmetric supercapacitor applications. Chem. Eng. J. 355, 881–890 (2019). https://doi.org/10.1016/j.cej.2018.08.185
V. Shobana, K. Balakrishnan, P. Parthiban, S. Angaiah, Electrospun Nd3+ doped LiMn2O4 nanofibers as high performance cathode materials for Li-ion capacitors. ChemElectroChem 4, 2059–2067 (2017). https://doi.org/10.1002/celc.201700161
A. Subasri, K. Balakrishnan, E.R. Nagarajan, V. Devadoss, A. Subramania, Development of 2D La(OH)3 /graphene nanohybrid by a facile solvothermal reduction process for high-performance supercapacitors. Electrochim. Acta 281, 329–337 (2018). https://doi.org/10.1016/j.electacta.2018.05.142
H.R. Naderi, P. Norouzi, M.R. Ganjali, H.G. Ranjbar, Synthesis of a novel magnetite/nitrogen-doped reduced graphene oxide nanocomposite as high performance supercapacitor. Powder Technol. 302, 298–308 (2016). https://doi.org/10.1016/j.powtec.2016.08.054
Acknowledgement
We gratefully acknowledge Iran University of Science and Technology for providing materials and facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Roshani, R., Tadjarodi, A. Synthesis of ZnFe2O4 nanoparticles with high specific surface area for high-performance supercapacitor. J Mater Sci: Mater Electron 31, 23025–23036 (2020). https://doi.org/10.1007/s10854-020-04830-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-04830-5