
Abstract
Tailoring effects of Ca-doping concentration on the structural, optical, and electrical properties of Ba1−xCaxTiO3 (x = 0.0 to 0.15) nanomaterials prepared via low-temperature sol–gel auto combustion technique are presented. XRD results reveal that all the prepared nanomaterials are crystallized in tetragonal structure with space group P4mm. FT-IR measurements are carried out in order to reveal the vibrational frequencies of bonds in the crystal lattice. A strong absorption peak due to Ti–O bond (TiO6 octahedron) appears at 538 cm−1 that shifts to a higher frequency with an increase in the doping of Ca ions. It is explained by the Coulomb interaction. Optical band gap of the sample increases with increase Ca doping as 3.44 eV (x = 0.0), 3.48 eV (x = 0.10), and 3.45 eV (x = 0.15). The BCT materials with x = 0.10 apparently show that the particle with short-range order can strongly influence the formation of intermediate states in the gap. Dielectric behavior is found governed by the interfacial polarization, whereas the ac conductivity supports the non-Debye process [n < 1] that is described using Jonscher’s power law. The conduction mechanism is found changing from small polaron to large polaron hopping as the Ca ions increase. Moreover, the electrical conductivity arising due to the movement of charged ions is correlated with the hopping and the lattice defects. The temperature-dependent activation energy is obtained in the range of 0.19–0.12 eV, which evidences the NTCR behavior observed for all studied materials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Perovskite type (ABO3)–BaTiO3 oxide materials are having their physical properties such that they find various applications in ferroelectric memories as surface acoustic wave device, micro-electromechanical systems, tunable microwave filters, and microwave capacitors [1]. In the last few decades, the optical and electrical properties of some relaxor ferroelectrics have been widely interrogated for application in wireless communication, metal–oxide–semiconductor, microwave dielectric, and field-effect transistor (FET) [2,3,4,5,6]. Barium titanate (BaTiO3) is an industrially important compound that exhibits ferroelectric properties at and above room temperature. It has five different crystallographic forms: hexagonal, cubic, tetragonal, orthorhombic, and rhombohedral. The hexagonal and cubic structures are paraelectric in nature while tetragonal, orthorhombic, and rhombohedral show ferroelectric behavior in nature [7,8,9]. The research for a suitable photo-electrode and photo-catalyst, perovskite-based metal oxides (ABO3) has been the subject of extensive studies during the past two decades [10,11,12] because they offer a wide range of possibilities for tailoring their structure through cationic substitutions either at A or B sites in order to tune their optical and electrical properties [13]. In Ba1−xCaxTiO3 (BCT), a widely accepted view is that a random field-induced ferroelectric domain state may be responsible for the observation of relaxor behavior. Nevertheless, it is ensure even if Ca2+ ions can cause Ca2+–Vo centers to form dipoles and thus set up a local random electric field [14, 15]. For both fundamental and commercial importance, enormous efforts have been pursued to modify the dielectric and ferroelectric properties of BaTiO3-based materials by A-site substitution. It comes from alkaline-earth (Sr2+ or Mg2+) and rare-earth ions (La3+, etc.) and their cation size effects that cover up Ti off-centering hence the lower Curie temperature. On the other side, the effects of Ca-substitutions in BaTiO3 are entirely unique and increasing interest has been received recently [16]. In recent years, the sol–gel technique has gained interest in the area of processing of nanomaterials, microstructure because of its several advantages such as easier composition control, better homogeneity, low processing temperature, and low equipment cost [17]. For the present investigations, BaTiO3 nano materials have been synthesized by Ca2+ ions doping on A-site using the sol–gel technique through organic precursor route. Ca-substitution could be intriguingly sustain the Curie temperature while other alkaline metals indicate the stern size affects the sustainable Curie temperature. The broadened tetragonal phase in Ba1−xCaxTiO3 system suggested the additional displacement of Ca attributing this phenomenon to both the displacements of Ca and their influence on Ti off-centering. They discovered that Ca has its own ferroelectric displacement as well as influence on Ti off-centering, which alleviates the lattice strain effect, and afterward maintains the Curie temperature. These reports firstly interpret that Ca has a displacement strength effect on its own A-site and Ti off-centering. Along with this deliberation, Ca-substitution could be a desirable choice to retain both the exquisite macroscopic electrical properties and room temperature dielectrics in BaTiO3-based materials. Therefore, the effects of Ca-substitution on the overall ferroelectric properties such as remanent polarization, coercive field, and temperature dependence of polarization in BaTiO3-based materials should be a very important issue. In this paper, we discuss the optical energy bandgap and ferroelectric characteristics in BaTiO3 materials that have been achieved by Ca2+ substitution on A-site.
2 Experimental procedure
2.1 Required materials
Barium (II) nitrate (Ba(NO3)2, Merck-99.0%), titanium (IV) butoxide (C16H36O4–Ti, Aldrich-97%), calcium (II) nitrate (Ca(NO3)2, Merck-98.0%), citric acid (C6H8O7), ammonia solution (NH4OH), and nitric acid (HNO3) were used to prepare all the required materials used in the present experiments.
2.2 Synthesis of BaTiO3 and Ca ion-modified BaTiO3 nanomaterials
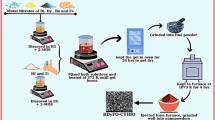
BaTiO3 and Ca ion-modified BaTiO3 nanomaterials were synthesized via a low-temperature sol–gel method as follows. Stoichiometric amounts of barium nitrate, titanium (IV) butoxide, and calcium nitrate as metal oxide precursors, and citric acid as a chelating agent were used as raw materials. These metal nitrates were homogeneously mixed in a molar ratio of Ba(NO3)2:Ti(C16H36O4):C6H8O7 = 1:1:2 in double-distilled water. Ammonia (NH4OH) solution was drop wise added to the above solution while stirring using the magnetic stirrer. The prepared homogeneous solution was maintained at pH ~ 7 with the help of litmus paper. The resulting solution was heated to 80 °C for 2 h to obtain a very viscous gel. After that increasing the heating temperature up to 100 °C leads to the ignition of gel form. The gel was dried in a hot air oven at 160 °C for 3 h. The dried precursor was obtained and ground to fine nanocrystalline powders then calcined at 900 °C for 2 h. For Ca substituted BaTiO3 catalysts, a similar procedure was followed with the stoichiometric addition of calcium nitrate to the above solution in the following ratio (1 − x)Ba(NO3)2:xCa(NO3)2:Ti(C16H36O4):C6H8O7 where x = 0.0 < x < 0.15, respectively. For pelletization, polyvinyl alcohol (PVA) was added in calcined powder and pressed into a circular disk of 1 mm thickness and 15 mm diameter by applying a pressure of ~ 6 tons. Sintering of the pellets was done at 1100 °C for 2 h.
2.3 Characterization technique
The prepared nanocrystalline powder has been characterized at room temperature. X-ray diffraction (XRD) patterns of the materials were recorded using a Bruker D8 advance diffractometer using Cu Kα (wavelength λ = 1.5406 Å) as the radiation source. The diffractometer uses a 1-D position-sensitive detector (Lynx Eye) based on the silicon drift detector technique, which reduces the measurement time significantly without any reduction in the diffracted intensity. Rietveld refinement analysis was performed in a step size 0.02 with 2θ varying from 20° to 80°. The structural parameters were obtained by using the Full Prof Suite program. The FT-IR spectra were recorded using the Frontier Perkin–Elmer (SP 10 STD) based on attenuated total reflectance (ATR) FT-IR spectrometer. A powerful sampling technique is given by the ATR system that removes sample preparation. The FT-IR spectra can be measured in the frequency range of 4000–400 cm−1. The optical absorption spectra were recorded by UV–Vis spectrometers using a Perkin–Elmer, USA Model: Lambda 950 having scanning double beam, double monochromator, and ratio recording spectrometer over the range of 200–800 nm and BaSO4 were used for calibration before the test and as a reference sample. The dielectric properties of the prepared samples were assessed with frequency at room temperature (RT) using a 6500B series Wayne Kerr precision impedance analyzer and Novo Control impedance analyzer. The experimental data were collected using software that allows power, dielectric constant, and dielectric losses to be acquired directly over a range of frequencies (20 Hz to 120 MHz).
3 Results and discussion
3.1 Phase and structure analysis
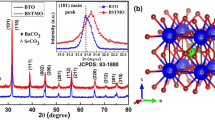
BaTiO3 and Ca ions-modified BaTiO3 powders synthesized using sol–gel chemical methods have been characterized by the XRD technique. The XRD patterns of the BTO and Ca-BTO indicate about the tetragonal structure with space group P4mm (99) PCPDF (file no. 82–2234). Figure 1a shows the diffraction peaks of pure and doped Ca ions with different concentration (x = 0.10, 0.15). The intense diffraction peaks are observed at 2θ = 22.17 (100), 31.63 (110), 38.87 (111), 45.20 (200/002), 50.86 (210), and 56.28 (211), respectively. The diffraction peaks of BTO shift towards lower angle 2θ after Ca doping [18]. Even for the concentration of x = 0.10, an orthorhombic phase of barium carbonate (BaCO3, PCPDF 85–0720) was noted, as shown in Fig. 1a. The formation of carbonates in the samples may have resulted from atmospheric contamination or, less likely, due to the presence of the carbon precursor titanium (IV) butoxide (C16H36O4–Ti). X-ray patterns were analyzed for lattice parameters (a, c), unit cell c/a, and particle size. The lattice parameter (a, c) value of the pure and Ca-doped materials was carefully determined by the Full Prof program as shown in Table 1. The tetragonality factors (c/a ratio) of the materials are slightly increased caused by Ca doping where as the unit cell volume of the BTO and Ca-BTO samples decreases because of the smaller ionic radius of Ca2+ (1.34 Å) than that of Ba2+ (1.61 Å). The average crystalline size (t) is determined by using the Debye–Scherrer’s formula and the accurate values of average crystalline size (D) is estimated using Williamson–Hall relation as given below:

a XRD pattern of Ba1−xCaxTiO3 (x = 0.0, 0.10, 0.15) materials. b Lattice strain—size analysis of 0% BT, 10% Ca-BT, and 15% Ca-BT materials. c Rietveld Refinements of Ba1−xCaxTiO3 material. The black open circles represent the observed pattern, The red color lines represent the calculated pattern, the difference between the observed and calculated profiles is represented by the blue color and green color represents the Bragg position
where k is the shape factor having value for the spherical particle as ~ 0.9, ɛ is the lattice micro-strain due to imperfection in crystals, and λ is the wavelength (0.154 nm) [19, 20]. Figure 1b shows the plot of βCosθhkl vs. 4Sinθhkl, where the slope is fitted by linear regression line that directly gives the value of lattice micro-strain (ɛ) according to the Williamson–Hall method and is given in Table 1. The W–H plot (Fig. 1b) shows a positive slope which indicates the presence of tensile strain at the lattice site. The accurate crystalline size (D) is also obtained by the intercept of the fitted linear regression line as follows:
The powder XRD pattern has been analyzed and their Rietveld refinements are performed for synthesized nanomaterials to estimate the changes in crystalline parameters as a result of the Ca-modified system. The powder XRD pattern along with Rietveld refined parameters using the Full Prof program is shown in Fig. 1c. The powder diffraction pattern is one of the most exciting tools to obtain qualitative and crystal structure information. XRD can be used not only for qualitative identification of polymeric forms of some multi-elemental ceramics but also for quantitative estimation of the different Bragg positions of various crystalline phases, which makes traditional X-ray diffraction pattern unsatisfactory. To resolve these problems, the Reitveld refinement method which is probably the most accurate and reliable technique to simulate the whole-pattern by least-square fitting refinement technique and has been successfully used for quantification and characterization of inorganic and organic compounds. Various profile fitting (R factors) of the experimental data such as Rp (profile factor), Rwp (weighted profile factor), and Rexp (expected weighted profile factor) makes it very prominent to conclude exact crystal structure symmetry which is defined as follows [21].
where Wi is the weight parameter, Iio, and Iic is the observed and calculated intensity for 2θi diffraction angle, (n–p)—number of degree of freedom, here ‘n’ is the total number of experimental points and ‘p’ is the number of refined parameters. The patterns are refined for the lattice parameter, background, Scale factor, Pseudo-Voigt profile factor (u, v, w), atomic (Wyckoff) position, and isothermal temperature factors (Biso). In tetragonal pure and modified BTO structure, the Ba atoms are in the Wyckoff position Ba at 1a (0, 0, 0), Ca at 1a (0, 0, 0), Ti at 1b (0.5, 0.5, 0.5224), O1 at 1b (0.5, 0.5, 0.0326), and O2 at 2c (0.5, 0, 0.4895). The fitted refined XRD patterns are in good agreement with the respective experimental data, noted by Rp, Rwp, Rexp, Bragg-R factor, and RF factor, which are listed in Table 2.
3.2 FT-IR analysis
The FT-IR spectra of the Ba1−xCaxTiO3 (x = 0.0, 0.10, 0.15) nanomaterials are shown in Fig. 2. A strong absorption peak associated with the vibrational frequency appears nearly at 538 cm−1. This absorption peak correlates to the asymmetric and symmetric stretching of the Ti–O (TiO6) octahedron bonds, highlighting with its lattice deformation. We detected a broadening and slight shift in this band to higher wavenumber, implying that the stretch of the Ti–O bond of BaTiO3 led to the corresponding mode of Ba1−xCaxTiO3 (x = 0.10, 0.15). It is explained by the weakness of Coulomb interaction [22], because the electric charge of cation Ca2+ is less than that of Ti4+. The electrostatic Coulomb force between Ca2+ and O2− is weaker than that between Ti4+ and O2−, which results in decreased energy of the M–O (cation—oxygen) bond. Another reason of the shift may be the replacement of Ba2+ by Ca2+, which decreases the cell volume of material because the ionic radii of Ca2+ (1.34 Å) is less than Ba2+ (1.61 Å). The volume of the direct cell parameter is decreased by doping also confirmed by the XRD patterns of Ba1−xCaxTiO3 [Table 1]. The direct cell volume is decreased and the resultant increase in the electrostatic Coulomb force interaction due to the shrinkage (order–disorder) of the Ti–O octahedron distance, the wavenumber of absorption becomes larger [23]. The main bands specific to the ionic NO3− (853 cm−1) introduced in the system by the catalyst barium, calcium, and titanium source as Ba(NO3)2, Ca(NO3)2, and TiO(NO3)2 in the case of the doped precursors are also identified. Other strong absorption bands at around 1425 cm−1, 1431 cm−1, and 1443 cm−1 are well defined in BCT materials (x = 0.0, 0.10, 0.15) that denoted the characteristic of the presence of carbonates. This band can be represented as the C=O vibration. In FT-IR spectra, we observe the presence of sharp bands around 1639 cm−1, 1752 cm−1, and 1758 cm−1 for all the samples which is related to the presence of O–H bonds. Corresponding to the OH− groups and water of hydration (adsorption to the particle surface), some bands appear around 1020 and 3525 cm−1, respectively. These are common in the systems that have hydrogen bonding, which also indicate about the defects caused by OH− groups in the titanate lattice deformation [24,25,26]. The strong absorption band at around 1431 cm−1 is well defined in materials having Ca2+ ion (x = 0.10) and is the characteristic of the presence of carbonate peaks, which is confirmed by the XRD measurement results.

FT-IR spectra of the Ba1−xCaxTiO3 samples: ax = 0, bx = 0.10, cx = 0.15
3.3 UV–Vis analysis
Figure 3a shows the UV–Vis absorption spectra of Ca-modified materials. Here, n electron in a non-bonding orbital is excited to the corresponding anti-bonding orbital π*, due to which the compounds containing double bonds like C=O undergoes n → π* transitions. These electronic transitions require minimum energy and show absorption at a wavelength above 200 nm (weak peak) which is accessible to UV–Vis spectroscopy. From Fig. 3a, the BC10T absorption peak is found at a higher wavelength compared to BTO and BC15T absorption. The other optical property of the BCT nanomaterials is determined by UV–Vis diffused reflectance spectra for materials having different concentrations of dopant at room temperature. The bandgap energy is one of the fundamental characteristics, particularly, when the optical devices are fabricated. When the material absorbs and scatters light, then the optical bandgap can be calculated from the diffuse reflectance spectroscopy. For powdered samples, the Kubelka – Munk (K–M) relation can be expressed as follows:

a UV–Vis absorption spectra of Ca-modified materials. b Tauc plot of BTO, BC10T, and BC15T materials. Inset illustration of a wide bandgap model composed of deep and shallow holes
where K is the K–M absorption coefficient and is directly proportional to the linear absorption coefficient (α), S is the scattering coefficient which is constant with the wavelength, and F(R) is called the K–M function. The optical bandgap Eg of the nanocrystalline material is determined from the well-known Tauc formula (Fig. 3b) when the K–M relation is fulfilled. The absorption of the material is proportional to F(R) and Tauc general formula can be expressed by:
where A is a constant and n is an electronic transition coefficient. Generally, n = 1/2 is for a direct allowed transition and n = 2 is for an indirect allowed transition. (F(R)hν)n is the modified K–M function. From Fig. 3b, it has been clearly seen that the UV–Vis spectra (Tauc plot) are a well-defined extrapolation of the linear fit. The optical bandgap, which indicates the presence of localized electronic state existing even for materials without substitution of the cation (Ca2+) at the A-site of perovskite BaTiO3 in the UV region has been obtained as an indirect wide bandgap (3.44 eV) [27]. The Ca2+-doped BTO materials show bandgap as 3.48 and 3.45 eV for x = 0.10 and 0.15, respectively (Fig. 3b). The optical bandgap in BCT-based oxide materials arises due to the transition from the O 2p orbitals (near the valence band) and Ca 3p orbitals as well as Ti 3d orbitals (below the conduction band). The observed values of the optical bandgap for ordered and disordered (lattice defects and impurities) materials indicate that the exponential absorption edge and the optical bandgap energy are controlled by the order–disorder structural degree in the lattice deformation under the effect of the octahedral crystal field of the six oxygen surrounding the titanates.
There are many quantum approaches that originated in low dimensional systems because the pairing and asymmetry energy plays an important role. In the atomic level, valence and conduction bands are associated with various degenerate energy levels. Density function theory (DFT) and band theory calculations suggest that these little bit energy states are also known as Fermi energy states. The typical energy gap in the Fermi level is ~ meV to eV range. Molecular orbital theory revealed s–p, p–d, and d–d transitions because the hybridization takes place between the covalent and ionic bonds. In the present studies, Ca-modified BTO-based nanomaterials show the charge transfer conduction, which is based on the electron–phonon coupling mechanism that has been explained on the groundwork of the Frank Condon principle. Oxide-based perovskite structure (BaTiO3) originates charge transfer between Ti4+ and O2− ions. In atomic spectra, the clouds of electrons are present in different sub-shells. Unpaired electrons are correlated with 2p and 3d orbit of O2− and Ti4+ ions, respectively.
Figure 3b illustrates the indirect wide bandgap present in the Ca-modified BTO-based nanomaterials. Indirect band anomaly occurs due to the electron transit from valence band maxima to the conduction band minima with the cooperation of photons and phonons. The maxima and minima of bands are located in a different position because the wave vector differs. The excited electron of the highest conduction band jumps to the low degenerate energy states in the conduction band because an excited electron releases some energy in very less time (~ 10–6 s). These electronic bands are associated with many vibrational levels. In the process of electron–phonon interaction, some phonons absorb or emit a very less amount of energy. Earlier reported results confirm that the Ca2+/Ti4+ ions show a strong contribution to the formation of Fermi energy levels near the conduction band [28, 29]. Henceforth, the energy gap little bit depends on the charge transfer conduction mechanism and this conduction phenomenon is mainly due to electron–phonon coupling.
The values of optical bandgap energy with increasing small amounts of concentration of the Ca2+ cation as observed in the spectra may be attributed to the way these intermediate states are situated within the bandgap. The effect of local crystalline distortion associated with the lattice defects or oxygen deficiency is also very common in perovskite materials. The sample with x = 0.10 has two absorption regions. The calculation of the band gap is made considering only the higher wavelength absorption region [30, 31]. The XRD data of this sample also indicate the presence of a carbonate peak without long-range order, and thus, the BCT sample with x = 0.10 shows particles with short-range order as well as a particle with long-range order (a regular shape of the particle). The sample with x = 0.15 shows a bandgap energy of approximately 3.45 eV, which is very close to BTO. Suppose the phase of BCT at x = 0.0 and 0.15 were not identified by XRD and FT-IR data, for this reason, it is evident that the value attained for the energy bandgap of this sample correlates to the band gap of the BCT compound and not to the energy gap of carbonates peak (single-phase identified by XRD), which has other optical absorption regions. The presence of a phase with short-range order can strongly influence the formation of intermediate states in the gap. According to the literature, a good understanding of the structurally disordered materials demonstrates a smooth continuous increase in the absorption as a function of energy, and this substantiates the behavior of UV–Vis absorption in this sample.
3.4 Dielectric study
The complex dielectric permittivity (real part (ɛ′) and imaginary part (ɛʺ)) as a function of frequency for the Ba1−xCaxTiO3 materials sintered at 11000C are illustrated in Fig. 4a. Dielectric constants of the Ca2+-doped BTO were calculated with different frequencies and different concentrations in the frequency regimes 10 Hz–1 MHz. It is observed that in Ba1−xCaxTiO3 materials, the dielectric constant values increase with a change in the concentration of Ca2+. For all the synthesized materials, a decrease in dielectric permittivity (ɛ′ and ɛʺ) with an increase in applied frequency is observed that reaches an almost constant value at a higher frequency that is expected due to Debye-type dielectric dispersion behavior which typically occurs in material. The obtained dielectric constant for BaTiO3 and Ba1−xCaxTiO3 (x = 0.10, 0.15) is 1490, 2188, and 1917, respectively, at a lower frequency (10 Hz). The dielectric constant of Ca2+-modified BTO systems is higher than the undoped BTO system.

a Frequency-dependant dielectric constant of real part (ɛ′), inset: imaginary part (ɛʺ) of dielectric constant of Ba1−xCaxTiO3 (x = 0, 0.10, 0.15). b Dielectric loss (tanδ) of Ba1−xCaxTiO3 (x = 0, 0.10, 0.15)
The result displays that the highest dielectric constant value is noticed for Ca doping with x = 0.10 in BTO. The dielectric constant value is much higher in lower frequency region, because in this region, atomic, ionic, interfacial (space charge), and electronic types of polarization play important role, but in higher frequency region, only electronic polarization affects the constant value [32]. At lower frequency regions, these charges have enough time to travel long distances in the materials and creating larger electronic polarization to achieve the dielectric constant of higher values. As the frequency increases, the charge carrier ions (electron hopping) cannot follow the applied external alternating field variations, which cause the dielectric constant (ɛ′) decrease indicating towards interfacial polarization, thereby resulting in a low dielectric constant due to decreases in polarizability. This dielectric dispersion behavior can be explained on the basis of the Maxwell–Wagner model of interfacial polarization with Koop’s phenomenological theory. The variation in dielectric constant with Ca2+ concentration can be explained on the basis of their lattice parameter ratio (c/a). Among these two synthesized Ca2+ doped BTO, x = 0.10 shows the highest c/a ratio, which resulted in a higher value of dielectric constant.
Energy loss (tan δ) is usually referred as dissipation factor in dielectrics. It is the ratio of εʺ (imaginary part) to the ε′ (real part) [tan δ = εʺ/ε′]. Figure 4b illustrates the loss tangent (tan δ) response of Ca-modified BTO nanomaterials with optimal doping (x = 0.10 and 0.15). The response of dielectric loss decreases as the frequency increases up to 1 MHz. The nature of dielectric loss peak attributes a stable relationship between the dielectric evolution and the conduction mechanism. Energy loss also depends on various factors such as grain and grain boundary contribution, defects, and methods adopted in the synthesis route [33]. In low-frequency region (10 Hz–1 kHz), a hump-like peak is observed in loss behavior, which is mainly due to a frequency resonance effect. In resonance condition, fR = 1/6.28τ where fR is the resonance frequency and τ is the relaxation time of charge carriers. Whenever the applied frequency is close to the hopping frequency of charge carriers (f = fR), the maximum amount of electrical energy is transferred to the oscillating ions (Ti4+). Henceforth, the peak appearance in dielectric loss reveals the higher amount of energy transfer in Ca-modified BTO nanomaterials.
When the optimal doping increases from x = 0.10 to 0.15, the dielectric loss also increases from 0.45 to ~ 0.86 as compared to the pristine BaTiO3 (~ 0.3) observed at 10 Hz. On the other hand, the resonance peak attributed to dielectric loss shifts towards the lower frequency as the doping concentration of Ca2+ ions increases. The observed resonance frequency (fR) is ~ 298 Hz, ~ 178 Hz, and ~ 32 Hz for BaTiO3 (x = 0), Ba0.9Ca0.1TiO3 (x = 0.10), and Ba0.85Ca0.15TiO3 (x = 0.15), respectively. This shifting of resonance peak also indicates the hopping frequency of charge carriers, which is mainly due to the hopping length of the octahedral site. Ca-modified BTO-based nanomaterials improve the capacity of charge carriers (dielectric constant) as well as overcome the energy loss. The frequency-dependent circuit requires a low value of the dielectric loss. Henceforth, these nanomaterials are permissible for fabrication point of view in low dimensional systems.
3.5 Electrical conductivity
In order to study the electrical transport behavior of modified materials, the samples were further characterized by ac conductivity (σac) variation with frequency at 300 K to understand the mechanism of conduction. Discussing the nature of ac conduction, Jonscher proposed the following universal power law:
here σdc is frequency-independent conductivity; Aωn denotes the transport behavior of charge carriers (polaron, electron, and ion) within the polycrystalline materials, A is the dispersion factor representing the strength of polarizability, ω is typically assigned to the hopping conduction. Finally, ac conductivity has a power-law behavior in terms of frequency where n is the frequency exponent value found in between 0 and 1 [34]. In Debye condition, n = 1, and in non-Debye condition , n > 1, which shows that the hopping motion is localized and n ≤ 1 shows that the motion is translational with sudden hopping. The frequency dependence of ac conductivity (σac) for all the materials is shown in Fig. 5a, which shows two distinct regimes within the measured frequency, (i) plateau, and (ii) the dispersion. The plateau regime corresponds to the frequency-independent conductivity σdc. The dc conductivity value is obtained at lower frequency. On the other side, dispersion regime is observed at a higher frequency.

a Non-linear fitting of ac conductivity (σac) and logarithmic σac with frequency dependence of Ba1−xCaxTiO3 (x = 0.0, 0.15). Inset: x = 0.10 of Ba1−xCaxTiO3. b Frequency dependence of ac conductivity at various temperatures for Ba1−xCaxTiO3 (x = 0.0 and 0.10) materials. c Variation of the frequency exponent value ‘n’ with temperature. d Arrhenius plot of Ba1−xCaxTiO3 (x = 0.0, 0.10, and 0.15) at 100 Hz
According to this dispersion, the σac shows a strong dependence on the frequency and increases linearly at high frequency. The conductivity vs. frequency data is fitted using power law and the various fitting parameters are obtained that is tabulated in Table 3. σdc was found to be the order of 10–8 S/cm to 10–6 S/cm, leading to the insulating behavior of the materials. The values of n have been found as 0.88 (x = 0.0), 0.59 (x = 0.10), and 0.86 (x = 0.15). The value of n < 1 signifies that the charge carriers (non-Debye behavior) present in the materials adopt a translational motion with sudden hopping [35, 36]. The observed dc conductivity behavior of Ca2+ ions partial substitution (x = 0.10) in BaTiO3 material is maximum that might be understood in terms of their structural defects or lattice parameters ratio of non-stoichiometric donor doped barium titanate. The Ba sites are generally occupied by Ca ions due to an electric charge and ionic radii and are described by the Kroger–Vink notation [37]:
where \({\text{Ca}} \cdot_{\text{Ba}}\) is ionized Ca donor dopants, \({2O}_{\mathrm{o}}^{X}\) represents the neutral oxygen atom. The Ca ions situated at A-site of BaTiO3 lattice yields a positive charge that can be reimbursed by ionic radii of the atoms.
Figure 5b elucidates the temperature dependence of ac conductivity behavior. A representative plot of the conductivity spectra of the Ba1−xCaxTiO3 (where x = 0.0 and 0.10) system at some selected temperature ranges is shown in Fig. 5b. The frequency exponent factor and temperature-dependent ‘n’ are the most important parameters used to illustrate the applicable models for the mechanism of conduction. It is observed (Fig. 5c) that the x = 0.0 parameter ‘n’ shows an increasing trend as a function of temperature then conduction mechanics is attributed to the small polaron hopping (non-overlapping) [38]. In contrast with Ca (x = 0.10)-substituted BTO samples, firstly the value of n decreases gradually and attains a minimum value at a particular temperature and afterward increases with further increase in temperature, which indicates to large polaron hopping (overlapping). It is clearly seen that the minimum value of ‘n’ corroborates the transition temperature (Tc), which shows that there is a maximum interaction between the charge carriers at Tc [39]. This is an indication of the evolution of large polarons and can be illustrated on the basis of overlapping large polaron hopping model. This implies the transition from small polaron to large polaron hopping in conduction mechanism with the substitution of Ca.
Figure 5d represents the Arrhenius plot of dc conductivity obtained from the linear fitting of the ac conductivity spectra with the composition up to 0.0 \(\le \) Ca \(\le \) 0.15. The conductivity of the material is thermally approved concept that follows Arrhenius law described as follows:
where σ0 denotes pre-exponential factor, Ea is the activation energy required to activate the mobile charge carriers, KB is the Boltzmann constant, and T is the absolute temperature. The reciprocal temperature dependence of dc conductivity for all the materials with linear fitting shows that the scale of conductivity decreases with increasing temperature [40]. The calculated values of the activation energy for all the materials are listed in Table 4. The conductivity of the BCT systems is described as the sum of electronic and ionic contributions as given below:
where ne and ni denotes the concentration of electrons and ions, µe and µi denotes the electronic and ionic mobility, q denotes the elementary charge of the particle. The values of activation energy of Ba1−xCaxTiO3 (x = 0.0, 0.10, and 0.15) at 100 Hz decrease in region-I as compared with region-II due to hopping charge carriers related to the oxidation and reduction processes [41]. At higher-temperature activation energy for (x = 0.1), the BCT sample decreases with increasing donor concentration, which arises the interaction between electron or hole that enhances due to non-stoichiometric defect chemistry or lattice deformation.
4 Conclusion
In summary, Ca-modified BaTiO3-based nanomaterials were successfully prepared by low-temperature sol–gel auto combustion technique. Laboratory X-ray diffraction and Rietveld refinement studies confirm that all the prepared nanomaterials have good crystalline nature. Structural analysis witness tetragonal crystal geometry with space group P4mm. FT-IR spectra reveal the vibrational bond of chemical ions present in the nanomaterials. Doping of Ca2+ ion in the place of Ba2+ ions decreases the cell volume, which is due to small ionic radii of Ca2+.
UV–Vis spectroscopy recorded in diffuse reflectance mode shows the electronic transition (n → π*) in the wavelength range > 200 nm. Indirect energy bandgap lies between ~ 3.44 and 3.48 eV, which indicates the semi-conducting behavior of the materials. Non-linear trends in band gap values as a function of doping concentration follow the charge transfer conduction mechanism attributed to electron–phonon interaction.
Dielectric constant and dissipation factor (energy loss) both shows decreasing behavior with increase in frequency. In high frequency region (> 103 Hz), the value of dielectric constant is steady because space charge polarization can not follow the applied electric field. Frequency and temperature-dependent ac conductivity (σac) behavior is well explained using Jonscher’s power law, which provides various parameters related with ac conductivity. Conduction in material due to charge carriers is strongly correlated with polaron hopping mechanism (long/short range) present in Ca modified. Activation energy has been found to be ~ 0.12 to 0.19 eV, which is mainly attributed to the oxidation/reduction process in charge carriers. The inverse of temperature-dependent dc conductivity results indicates the negative temperature coefficient of resistance (NTCR) present in all the prepared nanomaterials. The characteristic features of NTCR and minimum value of energy loss (< 1) indicate that these nanomaterials can be widely useful in high frequency power transformer, actuator, and transducer.
References
Y. Fujimori, T. Nakamura, H. Takasu, Low-temperature crystallization of sol-gel-derived Pb(Zr, Ti)O3 thin films. Jpn. J. Appl. Phys. 38, 5346 (1999)
A.A. Bokov, Z.-G. Ye, Recent progress in relaxor ferroelectrics with perovskite structure. J. Mater. Sci. 41, 31–52 (2006)
M.T. Sebastian, Dielectric Materials for Wireless Communication, Chapter 3 (Elsevier, Amsterdam, 2008), p. p58
C.D. Dimitrakopoulos, I. Kymissis, S. Purushothaman, D.A. Neumayer, P.R. Duncombe, R.B. Laibowitz, Low-voltage, high-mobility pentacene transistors with solution-processed high dielectric constant insulators. Adv. Mater. 11, 1372 (1999)
J. Xu, W Menesklou and E Ivers-Tiff´ee, investigation of BZT thin films for tunable microwave applications. J. Eur. Ceram. Soc. 25, 2289 (2005)
T. Badapanda, S.K. Rout, L.S. Cavalcante, J.C. Sczancoski, S. Panigrahi, E. Longo, M. Siu Li, Optical and dielectric relaxor behavior of Ba(Zr0.25Ti0.75)O3 ceramic explained by means of distorted clusters. J. Phys. D: Appl. Phys. 42, 17 (2009)
L. Huang, Z. Jia, I. Kymissis, S. O’Brien, High K capacitors and OFET gate dielectrics from self-assembled BaTiO3 and (Ba, Sr)TiO3 nanocrystals in the superparaelectric limit. Adv. Funct. Mater. 20, 554 (2010)
F.A. Yildirim, C. Ucurum, R.R. Schliewe, W. Bauhofer, R.M. Meixner, H. Goebel, W. Krautschneider, Spin-cast composite gate insulation for low driving voltages and memory effect in organic field-effect transistors. Appl. Phys. Lett. 90, 083501 (2007)
A. Kholkin, A. Seifert, N. Setter, Electromechanical properties of sol-gel derived Ca-modified PbTiO3 films. Appl. Phys. Lett. 72, 3374 (1998)
F.W. Van Keuls, R.R. Romanofsky, N.D. Varaljav, F.A. Miranda, C.L. Canedy, S. Aggarwal, T. Venkatesan, R. Ramesh, A Ku-band gold/BaxSr1−xTiO3/laalo3 conductor/thin-film ferroelectric microstrip line phase shifter for room-temperature communications applications. Microwave Opt. Technol. Lett. 20, 53 (1999)
W. Defa, Y. Jinhua, K. Tetsuya, K. Takashi, Photophysical and photocatalytic properties of SrTiO3 doped with Cr cations on different sites. J. Phys. Chem. B 32, 15824–15830 (2006)
C.L. Freeman, J.A. Dawson, J.H. Harding, L.B. Ben, D.C. Sinclair, The influence of A-site rare earth ion size in controlling the curie temperature of Ba1−xRExTi1−x/4O3. Adv. Funct. Mater. 23, 491 (2013)
I. Levin, V. Krayzman, J.C. Woicik, Local-structure origins of the sustained curie temperature in (Ba, Ca)TiO3 ferroelectrics. Appl. Phys. Lett. 102, 162906 (2013)
S.N. Yun, X.L. Wang, Dielectric properties of Bismuth doped Ba1−xCaxTiO3 ceramics. Mater Lett. 60, 2211–2213 (2006)
J.G. Bendroz, K.A. Muller, Sr1−xCaxTiO3: an XY quantum ferroelectric with transition to randomness. Phys. Rev. Lett. 52, 2289–2292 (1984)
M. Acosta, N. Novak, V. Rojas, S. Patel, R. Vaish, J. Koruza, G.A. Rossetti, J. Rodel, BaTiO3-based piezoelectrics: fundamentals, current status, and perspectives. Appl. Phys. Rev. 4, 041305 (2017)
D. Fu, M. Itoh, S. Koshihara, T. Kosugi, S. Tsuneyuki, Anomalous phase diagram of ferroelectric (Ba, Ca)TiO3 single crystals with giant electromechanical response. Phys. Rev. Lett. 100, 227601 (2008)
L. Ju, T. Sabergharesou, K.G. Stamplecoskie, M. Hegde, T. Wang, N.A. Combe, H. Wu, P.V. Radovanovic, Interplay between size, composition, and phase transition of nanocrystalline Cr3+-doped BaTiO3 as a path to multiferroism in perovskite-type oxides. J. Am. Chem. Soc. 134, 1136–1146 (2011)
A.K. Zak, W.A. Majid, M.E. Abrishami, R. Yousefi, X-ray analysis of ZnO nanoparticles by Williamson–Hall and size–strain plot methods. Solid State Sci. 13, 251–256 (2011)
K. Venkateswarlu, A.C. Bose, N. Rameshbabu, X-ray peak broadening studies of nanocrystalline hydroxyapatite by Williamson–Hall analysis. Phys. B 405, 4256–4261 (2010)
R.G. Teller, J.F. Brazdil, R.K. Grasselli, J.D. Jorgensen, The structure of γ-bismuth molybdate, Bi2MoO6, by powder neutron diffraction. Acta Cryst. C40, 2001–2005 (1984)
C.H. Perry, B.N. Khanna, G. Rupprecht, Infrared studies of perovskite titanates. Phys. Rev. 105, A408 (1957)
X. Jin, D. Sun, Y. Zhang, J. Qian, Investigation on FTIR spectra of barium calcium titanate ceramics. J. Electroceram. 22(1), 285–290 (2009)
A.K. Rai, K.N. Rao, L.V. Kumar, K.D. Mandal, Synthesis and characterization of ultrafine barium calcium titanate, barium strontium titanate and Ba1−2xCaxSrxTiO3 (x = 0.05-0.10). J. Alloys Compd. 475, 316–320 (2009)
N. Baskaran, H. Chang, Thermo-Raman and dielectric constant studies of CaxBa1−xTiO3 ceramics. Mater. Chem. Phys. 77, 889–894 (2002)
L. Wang, H. Kang, D. Xue, C. Liu, Synthesis and characterization of Ba0.5Sr0.5TiO3 nanoparticles. J. Crys. Growth 311, 605–607 (2009)
V.M. Longo, A.T. Figueiredo, A. Lázaro, M.F. Gurgel, M.G.S. Costa, C.O. Paiva-Santos, J.A. Varela, E. Longo, V.R. Mastelaro, F.S. Vicente, A.C. Hernandes, R.W.A. Franco, Structural conditions that lead to photoluminescence emission in SrTiO3: an experimental and theoretical approach. J. Appl. Phys. 104, 023515-1–23611 (2008)
L.-D. Yuan, H.-X. Deng, S.-S. Li, J.-W. Luo, S.-H. Wei, Unified theory of the direct or indirect band gap nature of conventional semiconductors. Phys. Rev. B. 98, 245203 (2018)
G. Kotnana, S. Jammalamadaka, Band gap tuning and orbital mediated electron–phonon coupling in HoFe1−xCrxO3 (0 ≤ x ≤ 1). J. Appl. Phys. 118(12), 124101 (2015)
E. Orhan, F.M. Pontes, C.D. Pinheiro, T.M. Boschi, E.R. Leite, P.S. Pizani, A. Beltrán, J. Andrés, J.A. Varela, E. Longo, Origin of photoluminescence in SrTiO3: a combined experimental and theoretical study. J. Solid State Chem. 177, 3879–3885 (2004)
V.M. Longo, M.G.S. Costa, A.Z. Simões, I.L.V. Rosa, C.O. Paiva-Santos, J. Andrés, E. Longo, J.A. Varela, On the photoluminescence behavior of samarium-doped strontium titanate nanostructure under UV light. A structural and electronic understanding. Phys. Chem. Chem. Phys. 12, 7566–7579 (2010)
N.V. Dang, T.D. Thanh, L.V. Hong, V.D. Lam, T.-L. Phan, Structural, optical and magnetic properties of polycrystalline BaTi1−xFexO3 ceramics. J. Appl. Phys. 110, 043914 (2011)
P. Choudhary, D. Varshney, Elucidation of structural, vibrational and dielectric properties of transition metal (Co2+) doped spinel Mg-Zn chromites. J. Magn. Magn. Mater. 454, 274–288 (2018)
S. Gattu, K.S. Dasari, R.V. Kocharlakota, Structural and dielectric properties of Sn doped barium magnesium zirconium titanate perovskite ceramics. World J. Condens. Matter Phys. 5, 343–352 (2015)
A.P. Almond, A.R. West, R.J. Grant, Temperature dependence of the a.c. conductivity of Naβ-alumina. Solid State Commun. 44, 1277–1280 (1982)
P. Saxena, D. Varshney, Effect of d-block element substitution on structural and dielectric properties on iron cobaltite. J. Alloy Compd. 705, 320–326 (2017)
G. Catalan, J.F. Scott, Physics and applications of bismuth ferrite. Adv. Mater. 21, 2463 (2009)
F.A. Kröger, H.J. Vink, Relations between the concentrations of imperfections in crystalline solids. Solid State Phys. 3, 307–435 (1956)
S.R. Elliott, A.c. conduction in amorphous chalcogenide and pnictide semiconductors. Adv. Phys. 36, 135–218 (1987)
P. Singh, O. Prakash, D. Kumar, Electrical conduction behavior of perovskite oxide system BaSnNbO. Solid State Ion. 176, 2167–2170 (2005)
P. Singh, O. Prakash, D. Kumar, DC conduction behavior of niobium doped barium stannate. J. Mater. Sci. Mater. Electron. 16, 145–148 (2005)
Acknowledgments
The authors are thankful to Devi Ahilya university, Indore, for research fellowship (Golden Jubilee Fellowship) ref. no. Kra. Vikas/Swarna Jayanti Fellowship/2018/433 and acknowledge Dr. Mukul Gupta for XRD measurement, Dr. Uday Deshpande for FT-IR and UV–Vis measurements, and Mr. Bhardwaj for Dielectric measurement of UGC-DAE CSR, Indore.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bisen, S., Khan, M. & Mishra, A. Tailoring effect of large polaron hopping in the conduction mechanism of Ca-modified BaTiO3 system. J Mater Sci: Mater Electron 31, 9212–9223 (2020). https://doi.org/10.1007/s10854-020-03452-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-03452-1