
Abstract
In the present study, polypyrrole (PPy) and PPy/MnO2 composites were synthesized using a modified chemical oxidation polymerization method. The polymerization was carried out in the aqueous medium using FeCl3·6H2O as an oxidant. The synthesized materials were characterized using XRD, SEM, EDX, TGA, FT-IR, surface area analyzer, UV–visible spectrophotometry and LCR-meter. The results confirmed the successful synthesis of materials with amorphous and crystalline nature of PPy and PPy/MnO2 composites, respectively. PPy/MnO2 composites were found to be thermally more stable with large surface area as compared to PPy. The PPy was found to be porous in nature while PPy/MnO2 composites have compact structure. The elemental analysis confirmed the presence of MnO2 in the composites; however, greater quantity of MnO2 was observed than the added amount with an efficient influence on the doping level of PPy. The synthesized materials showed semiconducting nature and their resistance was found to be dependent on temperature and MnO2 content in the composites.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The synthesis and characterization of conducting polymers (CPs) have become an important area of research in polymer science and engineering due to their excellent electrical properties [1]. The CPs have conjugated structures consist of alternate single-double bonds which is one of the basic requirement for their electrical conductivity. During oxidation process, some of the π-bonds are broken, releasing electrons which further improve their conductivity [2]. These polymers have vast analytical, technological and commercial uses due to their greater electrical conductivity, thermal stability, biocompatibility, easy method of preparation and scale-up [3]. Owing to their unique characteristics, CPs have been extensively used in lithium batteries, LEDs, gas separation membranes, electro-chromic displays, sensors, etc. [4,5,6,7,8,9,10,11]. However, due to the rigid conjugated back bone structures, these polymers suffer from the problem of poor processibilty. Similarly, CPs are infusible because of their chemical stability and brittleness [12,13,14,15]. The poor mechanical strength of CPs also limits their usage in various fields [16]. To overcome such drawbacks, researchers have shown interest toward composites of CPs particularly with metal oxides. These composite materials have attractive physical, chemical, thermal and mechanical properties [17,18,19].
PPy is one of the most important CPs which has attracted the attention of many researchers. PPy can be synthesized easily by oxidation polymerization methods [20,21,22]. During oxidation process, some positive charges called polarons or bipolarons are introduced into the polymer back bone. The movements of these charges are mainly responsible for its greater conductivity; reaching some time to that of metals [23]. However, like other CPs, PPy also faces the aforementioned problems. In order to get rid of those shortcomings and to improve its characteristics, the composites of PPy particularly with metal oxides are preferred. For example, Jang et al. [24] studied the ammonia sensitivity of PPy/multi-walled carbon nanotube (MWCNT) composites. The performance of the materials was increased with MWCNT contents. Tandon et al. [25] studied the sensing behavior PPy–Fe3O4 nanocomposite for humidity and some other gases like CO2, N2 and O2. Due to good sensing behavior, the materials were declared to be used as gas sensor. Bhende et al. [26] prepared PPy/CuO nanocomposites by in situ polymerization of Py in the presence of varying amounts of CuO using FeCl3 as an oxidant. The electrical conductivity of the composites was observed to increase with increase in temperature up to 8 wt% of CuO; after than a decrease was observed. Yin et al. [27] synthesized CuO/PPy nanocomposite and studied their electrochemical properties. When assessed as anode material for lithium-ion batteries, the nanocomposite showed much better capacity and performance than bare CuO nano-belts.
MnO2 is an important semiconducting material which is widely used in electrical devices [28, 29]. Looking into the importance of MnO2, this metal oxide was selected for the preparation of PPy/MnO2 composites using a simple oxidation polymerization protocol [30,31,32] with little amendments, aiming to achieve composite materials of optimized structure and better electrical characteristics. The structural, thermal and electrical properties of the synthesized materials were investigated.
2 Experimental
2.1 Chemicals
The Py, FeCl3·6H2O (oxidant), MnO2, ethanol and acetone were of analytical grade (Merck, Germany). The Py was stored after distillation under reduced pressure. The required solutions of Py and FeCl3·6H2O were prepared in deionized double distilled water (DDDW).
2.2 Synthesis of PPy and PPy/MnO2 composites
For the synthesis of PPy, 1 mL of 1 M Py aqueous solution was added slowly into a 10 mL of 0.1 M aqueous solution of FeCl3·6H2O under magnetic stirring and N2 flow at 8 °C for 2 h. After completion of the reaction, a black precipitate of PPy produced which was filtered by vacuum filtration and rinsed with copious amount of DDDW, ethanol and acetone to remove unreacted reactants, oligomers, etc. The obtained PPy was dried at 60 °C for 4 h in an oven (LDO-030E, Daihan Lab Tech Co., Ltd., South Korea). For the synthesis of composites, 0.2 g MnO2 was dispersed in 10 mL of 0.1 M aqueous FeCl3.6H2O solution and sonicated at 25 °C for 30 min; followed by the addition of 1 mL of 1 M Py aqueous solution under the same conditions as mentioned for the synthesis of PPy. The resultant composite was named as PPy/2%MnO2. Similarly, other composites namely; PPy/5%MnO2, PPy/8%MnO2 and PPy/10%MnO2 were obtained under the same experimental conditions by adding 0.5, 0.8 and 1 g of MnO2, respectively. The schematic view of the experimental arrangement is shown in Fig. 1.

Schematic representation of the experimental setup
2.3 Characterization of synthesized materials
2.3.1 X-ray diffractometry (XRD)
XRD measurement of the synthesized materials was carried out using a JEOL X-ray diffractometry (JDX-3532-XRD Japan) with CuKα radiations, scanning range of 2θ of 10°–80°, scan speed of 0.1°/s and step angle of 0.05°.
2.3.2 Surface morphology and elemental analysis
The surface morphology and elemental composition of the synthesized materials was studied using SEM/EDX (JSM-5910, JOEL, Japan). During SEM analysis, the samples were attached to the stubs with the help of carbon conducting tape followed by sputtering with gold in fine coater (SPI-module). For EDX analysis, the samples were prepared by the same way but without sputtering with gold. The SEM analysis was carried out at 10 keV with 10 mm gap between the sample and electron gun tip while EDX spectra were obtained at 10 keV.
2.3.3 Thermal analysis
Thermal properties of the synthesized materials were investigated by thermogravimetric analyzer (Diamond TG/DTA, PerkinElmer, USA) at the temperature from 25 to 700 °C with the scanning rate of 10 °C/min under nitrogen atmosphere using alumina ceramic pan.
2.3.4 Fourier transform infrared (FT-IR) spectrometry
FT-IR spectrometry was used for the structural analysis of the synthesized materials. For this purpose, a Shimadzu FT-IR spectrometer (FTIR-8400, Japan) was used with wavenumber in the region of 4000–400 cm−1. An appropriate quantity of the sample was ground-mixed with KBr (IR grade) and a small quantity of this material was added to the sample holder of the diffuse reflectance accessory (DRS-8000 A) for FT-IR analysis at room temperature.
2.3.5 Surface area study
The surface area of the synthesized materials was obtained by multipoint Brunauer-Emmett-Teller (BET) method using surface area and pore size analyzer (Nova 2200 e, Quanta chrome, USA) via N2 sorption at 77 K. A known amount of the sample was placed in quartz tube and attached to the degassing station of the instrument at 90 °C for 3 h to remove the adsorbed moisture or any other volatile impurities. After degassing, the sample cell was attached to the analysis station to determine the multipoint BET surface area.
2.3.6 UV–visible spectrophotometry
UV–visible absorption spectrophotometry of the synthesized materials was carried out using a double beam UV–visible spectrophotometer (Lambda 650, PerkinElmer, USA) equipped with quartz cuvettes of 1 cm path length. The samples were dispersed in ethanol and spectral analyses were carried out from 200 to 800 nm wavelength.
2.3.7 Electrical characteristics
For the determination of electrical properties of the synthesized materials, the samples were deposited on the locally prepared sensor plate having interdigitated copper electrodes with a space of ~ 93 µm between the fingers. A suspension of each sample was prepared in acetone and then deposited on the sensor plate by spin-coating method at 3000 rpm for 1 min. For the removal of the vehicle acetone, the sample coated electrodes were heated in an oven at 60 °C for 4 h. The dimension of sensing part was 1.4 × 1.7 cm. The thickness and uniformity of the deposited layers was kept as same as possible. For the study of electrical resistance, the sensor plate was attached to LCR-meter (GW INSTEK LCR-817, Taiwan). For the study of temperature effect, the sensor plate in a glass chamber was placed inside a furnace. The variation of resistance of the samples with temperature was observed from 30 to 70 °C.
3 Results and discussion
3.1 XRD study
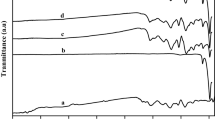
The XRD spectra of PPy, MnO2 and PPy/MnO2 composites are depicted in Fig. 2. A broad peak (Fig. 2a) at 2θ, 20°–30° indicates that PPy is of amorphous nature [33] and is because of the scattering from PPy chains interplanar spacing [34]. The approximate chain separation of PPy was determined from the maxima using the following relation [35,36,37].
where S is the chain separation of the polymer, λ is the wavelength of X-rays used and θ is the angle of diffraction at the maximum intensity of the amorphous halo. The average polymer chain separation of PPy was found to be 4.54 Å.

XRD patterns of (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2, (e) PPy/10%MnO2 and (f) MnO2
The Fig. 2f shows the crystalline nature of MnO2 and matches with ICCD PDF-4 Card # 04-004-9017. The characteristics peaks of MnO2 were found at 2θ = 28.68°, 37.39°, 41.01°, 42.85°, 56.70° and 59.404° which are assigned to (110), (101), (200), (111), (211) and (220) lattice planes, respectively. In case of XRD spectra of PPy/MnO2 composites (Fig. 2b–e), MnO2 reserves its crystallinity with no change in the peak position. However, the peak intensity was decreased which may be due to the relative increase of PPy contents.
The average crystallite size of pure MnO2 and PPy/MnO2 composites was determined using Debye–Scherrer equation [38,39,40];
In Eq. (2), L is the crystallite size, θ is the angle of diffraction, β is the full width at half maximum and k is the shape factor (0.89). The crystal size was determined using the intense peak at 2θ = 28.68° in case of all the samples. The crystal size of PPy/2%MnO2, PPy/5%MnO2, PPy/8%MnO2, PPy/10% MnO2 and MnO2 was found to be 17.89, 17.24, 16.45, 15.39 and 13.34 nm, respectively. Thus, the crystal size decreases with the increase of MnO2 content in the composites which may due to the crystalline nature of MnO2.
3.2 SEM and EDX analyses
SEM images of the synthesized materials are shown in Fig. 3. The results show that PPy is of hemispherical, globular and spongy shape. The porous nature represents a weak linkage among the polymeric particles. The approximate grain size of PPy is found to be 0.5 µm. In case of the composites, the particles are well connected. Since, the composites are gained by mixing MnO2 particles with Py and then polymerization carried out; therefore, the particles morphology of the PPy/MnO2 composites is nearly alike, i.e., elongated shape. The maintenance of the core particles, i.e., MnO2 morphology in the composites, suggests the development of a thin layer of polymer around MnO2 particles. Similar preservation of core-particles morphology in the composites has also been reported elsewhere [31, 32, 40]. The EDX spectrum of PPy/2%MnO2 is revealed in Fig. 4. The atomic weight percent of Mn, O, C, N and Cl are found to be 8.52, 27.84, 50.05, 8.74 and 4.86%, respectively in PPy/2%MnO2. Thus, the molar ratio of C, N, Cl and MnO2 is found to be 4.17, 0.62, 0.137 and 0.418, respectively. This shows that the actual amount of MnO2 in the composite is different from amount added. Similar results were also reported for of PPy/V2O5 composites [32]. These findings also supported the thermal analysis as discussed in Sect. 3.3.

SEM images of (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2 and (e) PPy/10%MnO2

EDX spectrum of PPy/2%MnO2
3.3 Thermal study
The TGA curves of PPy, MnO2 and PPy/MnO2 composites are displayed in Fig. 5. The results show that the pure MnO2 is stable up to 600 °C (Fig. 5f), while PPy and PPy/MnO2 composites display thermal degradation in the studied temperature range. The thermal properties of PPy are in-line with the reported data [32]. The thermal stability of the composites (Fig. 5b–e) increases with MnO2 contents due to the barrier effect of MnO2 and smaller movement of polymeric chains when attached to the metal oxide particles. In case of the composites, the residual weight increases with MnO2 contents. The approximate wt% of MnO2 in the composites without PPy was found to be 4, 53, 62 and 68% for PPy/2%MnO2, PPy/5%MnO2, PPy/8%MnO2 and PPy/10%MnO2, respectively. The TGA results show that the actual amount of MnO2 in the composites is different from the added percent weight. Since, we used relatively low concentration of the reactants (Py and FeCl3·6H2O), which resulted in the relatively smaller yield of PPy. Thus, the relative wt% of MnO2 in the composites is greater according to our expectations. The data clearly accomplish one of the main aims of study, which is to obtain composite materials with a thin layer of the polymer deposited on the metal oxide particles.

Thermograms of (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2, (e) PPy/10%MnO2 and (f) MnO2
In order to study the impact of MnO2 contents on the thermal stability, the activation energy of PPy and PPy/MnO2 composites decomposition was calculated by Broido method [41, 42] using the following equations;
where
In Eq. (4), Y is the fraction of undecomposed sample, Ea is the activation energy and R is the general gas constant. Similarly, in Eq. (5), wt is the sample weight at any time t, wf is the residual weight, w0 is the starting weight. The plots for activation energy calculation are displayed in Fig. 6 and the activation energy values were obtained from the slope of the plot of \(\ln \ln (1/Y)\) vs. \(1/T\) as shown in Table 1. The activation energy increases slightly with the increase of MnO2 content in the composites, i.e., from PPy to PPy/10%MnO2 and hence the thermal stability. Thus, the thermal study revealed that MnO2 particles have not a significant effect on the thermal stability of PPy.

Activation energy plots for (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2 and (e) PPy/10%MnO2
3.4 FT-IR spectroscopy
FT-IR spectra of PPy, MnO2 and PPy/MnO2 composites are presented in Fig. 7. The successful synthesis of PPy has already been reported in our previous studies [31, 32]. From the FT-IR spectrum of MnO2 (Fig. 7a), the bands at 705 and 564 cm−1 are respectively related to v6 and v8 vibration modes of MnO2 [43]. The FT-IR spectra (Fig. 7b–d) of the composites show all the bands of both PPy and MnO2 with no new bands. However, the band at 1536 cm−1 in PPy spectrum has shifted to 1570 cm−1 in the composites spectra which may be due to some sort of sensitive physical interaction between PPy and MnO2 particles. The FT-IR results suggest that there is no chemical interaction between PPy and MnO2 particles. Similar results were also reported for PPy/V5O2 composites [31, 32].

FT-IR spectra of (a) MnO2, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2, (e) PPy/10%MnO2 and (f) PPy
3.5 Surface area analysis
The surface area of the synthesized materials was determined by multipoint BET method as shown in Fig. 8. The plots are based on the following equation [44];
where W is the weight of adsorbed gas at a relative pressure P/P0, Wm is the weight of adsorbate constituting a monolayer of surface coverage and C is the BET C-constant.

BET plots for (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2 and (e) PPy/10%MnO2
The value of Wm can be obtained using the relation (6);
where s is the slope and i is the intercept of the plot obtained by plotting \(1/W(({P_0}/P) - 1)\) vs. \(P/{P_0}\) as shown in Fig. 8.
The BET surface area of the synthesized materials based on these plots for the linear part of the isotherms is presented in Table 1. The results show that the surface area of the synthesized materials increases with MnO2 contents. This may be due to the disperse nature of MnO2 particles. The effect of MnO2 contents on the microstructure morphology is clear from the SEM images (Fig. 3). The content of MnO2 in the composites has improved the porosity and hence surface area of the samples. All the materials exhibited adsorption isotherms with shape similar to type-II, according to the IUPAC classification [45]. From the isotherms (data not shown), it was concluded that the materials are of macroporous nature [46]. One of the important problems related to the applications of microporous (pore size of < 2 nm) and mesoporous (pore size of 2–50 nm) materials [47, 48] particularly in gas sensing devices is the intolerable slow diffusion of analyte gas inside the bulk of sensing material. Similarly, the adsorb gas molecules are removed with difficulty. This results in the unwanted long response and recovery time of the sensor. This issue can be easily resolved by the applications of macroporous materials (pore size of > 50 nm) [49] as in the present case.
3.6 UV–visible spectrophotometric study
In order to find out the effect of MnO2 particles on PPy doping level, UV–visible spectrophotometry of PPy and PPy/MnO2 composites was carried out from 200 to 800 nm wavelength as shown in Fig. 9. The UV–visible spectrum of PPy shows a distinct absorption around 280 nm which is credited to π–π* transition of benzenoid ring. The same peak was also observed in the absorption spectra of the composites with a red shift. The red shift can be related to the interaction between metal oxide particles and polymeric chains of PPy [50]. The band gap energy (Eg) of pure PPy and PPy/MnO2 composites was calculated using the relationship (7) [32, 51]:
where Eg is the band gap energy, h is Planks constant, C is the speed of light and λ is the cut-off wavelength. The values of Eg for PPy, PPy/2%MnO2, PPy/5%MnO2, PPy/8%MnO2 and PPy/10%MnO2 composites are 3.89, 3.69, 3.62, 3.52 and 3.41 eV, respectively. The smaller Eg value of the composites designates the relatively greater doping of PPy in the composites as compare to the pure PPy.

UV–visible spectra of (a) PPy, (b) PPy/2%MnO2, (c) PPy/5%MnO2, (d) PPy/8%MnO2 and (e) PPy/10%MnO2
4 Electrical properties
4.1 Effect of MnO2 contents on electrical resistance
Figure 10 displays the resistance of the synthesized materials at room temperature. PPy has the highest value of electrical resistance, i.e., 521 kΩ. By adding MnO2 (2 wt%), the resistance has dropped to 81 kΩ followed by slight decrease with increase in MnO2 contents evenly up to PP/10%MnO2. The higher electrical resistance of PPy may be due to the poor linkage among the polymeric chains [30] which decrease the movement of positive charge carriers. In the present study, higher value of the resistance shown by PPy compared to the reported data [52] can also be related to the relatively greater average polymer chain separation of PPy (4.54 Å) as revealed from the XRD results. On the other hand, all the composites have smaller electrical resistance which can be the result of several factors. The MnO2 particles have improved the conjugation and linking through the grain boundaries of PPy chains facilitating the charge carrier motion and decrease the resistance [26]. Similarly, the crystallinity of the composites also increases with MnO2 contents which further improve their conductivity. Another important reason for the smaller resistance of the composites is their relatively smaller band gap energy values as discussed earlier.

Variation of resistance of the synthesized materials with respect to MnO2 contents
4.2 Effect of temperature on the electrical resistance
The change in resistance of PPy and PPy/MnO2 composites with respect to temperature was studied from 25 to 70 °C and results are shown in Fig. 11. The resistance of all the samples decreases with the increase in temperature which confirms their thermally activated behavior. In case of PPy, the improvement in charge transfer efficiency with temperature decreases the resistance [53, 54]. The higher temperature increases the lattice vibration in PPy which cause chain stretching followed by the charge distribution all over the polymer chains. The ultimate result is the improvement in polymeric chains arrangement having ability for extended conjugation. It causes the intra and inter chain hoping of charge carriers which drops the resistance [55]. At higher temperature, there will be also molecular rearrangement in PPy, making the molecules suitable for efficient moment of charge carriers [56]. Similar behavior was also observed for the PPy composites contain semiconducting MnO2; where the resistance of the composites also decreases with temperature like pure MnO2 (Fig. 11). Similar trend was also reported in a previous study [31]. This again confirms the physical interaction between the PPy and MnO2 and declares the semiconducting nature of the synthesized materials [31, 57].

Variation of the samples resistance with temperature
5 Conclusions
In the present study, PPy and PPy/MnO2 composites were successfully synthesized in the aqueous medium via a modified chemical-oxidation polymerization method using low concentration of the reactants. The structural, thermal, morphological and electrical properties of the synthesized materials were investigated. The results confirmed the successful synthesis of materials with amorphous and crystalline nature of PPy and PPy/MnO2 composites, respectively. PPy/MnO2 composites were found to be thermally more stable with larger surface area as compared to PPy. Similarly, the PPy consisted of globular particles while the composites were of rod shape morphology. All the composites showed improved characteristics as compared to PPy. The electrical resistance of the composites was found to decreases with increase of MnO2 contents owing to the increase in their crystallinity and PPy doping. The materials were of semiconducting nature. The synthesized composites have all the characteristics of a typical sensing material [31] which is our future plan.
References
M.T. Ramesan, J. Appl. Polym. Sci. 128, 1540 (2013)
F. Kanwal, S.A. Siddiqi, A. Batool, M. Imran, W. Mushtaq, T. Jamil, Synth. Met. 161, 335 (2011)
H. Zengin, B. Erkan, Polym. Adv. Technol. 21, 216 (2010)
J.C. Vidal, E. Garcia, J.R. Castillo, Anal. Chem. Acta 385, 213 (1999)
T.E. Campbell, A.J. Hodgson, G.G. Wallace, Electroanalysis 11, 215 (1999)
D. Kincal, A. Kumar, A.D. Child, J.R. Reynolds, Synth. Met. 92, 53 (1998)
N.T. Kemp, G.U. Flanagan, A.B. Kaiser, H.J. Trodahl, B. Chapman, A.C. Partridge, R.G. Buckley, Synth. Met. 101, 434 (1999)
C. Jerome, D. Labaye, I. Bodart, R. Jerome, Synth. Met. 101, 3 (1999)
E. Smela, J. Micromech. Microeng. 9, 1 (1999)
G.S. Akundy, R. Rajagopalan, J.O. Iroh, J. Appl. Polym. Sci. 83, 1970 (2002)
T. Ito, P. Buehlmann, Y. Umezawa, Anal. Chem. 71, 1699 (1999)
X. Feng, Z. Sun, W. Hou, J.-J. Zhu, Nanotechnology 18, 195603 (2007)
J. Jiang, L. Ai, L. Li, J. Phys. Chem. B 113, 1376 (2009)
J. Jiang, L.H. Ai, J. Mater. Sci.: Mater. Electron. 21, 687 (2010)
H. Li, Y. Jia, S. Luan, Q. Xiang, C.C. Han, G. Mamtin, Y. Han, L. An, Polym. Compos. 29, 649 (2008)
A. Bhattacharaya, D.C. Mukherjee, J.M. Gohil, Y. Kumar, S. Kundu, Desalination 225, 366 (2008)
K. Majid, R. Tabassum, A.F. Shah, S. Ahmad, M.L. Singla, J. Mater. Sci.: Mater. Electron. 20, 958 (2009)
H. Yuvaraj, E.J. Park, Y.S. Gal, K.T. Lim, Colloids Surf. 313–314, 300 (2008)
M. Arain, A. Nafady, Sirajuddin, Z.H. Ibupoto, S.T.H. Sherazi, T. Shaikh, H. Khan, A. Alsalme, A. Niaz, M. Willander, RSC Adv. 6, 39001 (2016)
A. Joshi, S.A. Gangal, S.K. Gupta, Sens. Actuators B 156, 938 (2011)
H. Yoon, M. Chang, J. Jang, J. Phys. Chem. B 110, 14074 (2006)
D.U. Kim, B. Yoo, Sens. Actuators B 160, 1168 (2011)
C. Basavaraja, E.A. Jo, B.S. Kim, D.G. Kim, D.S. Huh, Macromol. Res. 18, 1037 (2010)
W.K. Jang, J. Yun, H. Kim, Y.S. Lee, Carbon Lett. 13, 88 (2002)
R.P. Tandon, M.R. Tripathy, A.K. Arora, S. Hotchandani, Sens. Actuators B 144, 768 (2006)
M.S. Bhende, S.P. Yawale, S.S. Yawale, Int. J. Innovat. Emerg. Res. Eng. 3, 488 (2016)
Z. Yin, Y. Ding, Q. Zheng, L. Guan, Electrochem. Commun. 20, 40 (2012)
M. Toupin, T. Brousse, D. Belanger, Chem. Mater. 14, 3946 (2002)
X. Wang, Y. Li, J. Am. Chem. Soc. 124, 2880 (2002)
A. Batool, F. Kanwal, M. Imran, T.J. Saadat, A. Siddiqi, Synth. Met. 161, 2753 (2012)
H. Khan, K. Malook, M. Shah, J. Mater. Sci.: Mater. Electron. 28, 13873 (2017)
K. Malook, H. Khan, M. Shah, Ihsan-Ul-Haque, Korean J. Chem. Eng. 35, 12 (2018)
N.S. Allena, K.S. Murray, R.J. Fleming, B.R. Saunders, Synth. Met. 87, 237 (1997)
J.Y. Ouyang, Y.F. Li, Polymer 38, 3997 (1997)
K. Cheah, M. Forsyth, V.T. Truong, Synth. Met. 101, 1 (1999)
P. Lemon, J. Haigh, Mater. Res. Bull. 34, 665 (1999)
K. Cheah, M. Forsyth, V.T. Truong, Synth. Met. 94, 215 (1998)
A. Aytimur, I. Uslu, S. Kocyigit, F. Ozcan, Ceram. Int. 38, 3851 (2012)
K.M. Parida, A.C. Pradhan, J. Das, N. Sahu, Mater. Chem. Phys. 113, 244 (2009)
H. Khan, A.K. Khalil, A. Khan, K. Saeed, N. Ali, Korean J. Chem. Eng. 33, 2802 (2016)
S. Gopalakrishnan, R. Sujatha, Der Chem. Sin. 2, 103 (2011)
P.S. Abthagir, R. Saraswathi, S. Sivakolunthu, Thermochim. Acta 411, 109 (2004)
C.M. Julien, M. Massot, C. Poinsignon, Spectrochim. Acta A 60, 689 (2004)
D.P. Lapham, J.L. Lapham, Int. J. Pharm. 530, 364 (2017)
Z.A. Alothman, Materials 5, 2874 (2012)
M. Thommes, K. Kaneko, A.V. Neimark, J.P. Olivier, F.R. Reinoso, J. Rouquerol, K.S.W. Sing, Pure Appl. Chem. 87, 1051 (2015)
H.P. Hentze, M. Antonietti, Rev. Mol. Biotechnol. 90, 27 (2002)
Y. Lumelsky, J. Zoldan, S. Levenberg, M.S. Silverstein, Macromolecules 41, 1469 (2008)
F.S. Macintyre, D.C. Sherrington, Macromolecules 37, 7628 (2004)
M. Alam, A.A. Ansari, M.R. Shaik, N.M. Alandis, Arab. J. Chem. 6, 341 (2013)
C. Piewnuan, J. Wootthikanokkhan, P. Ngaotrakanwiwat, V. Meeyoo, S. Chiarakorn, Superlattices Microstruct. 75, 105 (2014)
M.A. Chougule, S.G. Pawar, P.R. Godse, R.N. Mulika, S. Sen, V.B. Patil, Soft Nanosci. Lett. 1, 6 (2011)
M. Leclerc, G.D. Aparno, G. Zotti, Synth. Met. 55, 1527 (1993)
F. Zuo, M. Angelopoulos, A.G. MacDiarmid, A.J. Epstein, Phys. Rev. B 5, 3475 (1987)
M.A. Chougule, G.D. Khuspe, S. Sen, V.B. Patil, Appl. Nanosci. 3, 423 (2013)
A. Kobayashi, H. Ishikawa, K. Amano, M. Satoh, E. Hasegawa, J. Appl. Phys. 74, 296 (1993)
T. Fatima, T. Sankarappa, R. Ramanna, Int. J. Adv. Res. Phys. Sci. 3, 7 (2016)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khan, H., Malook, K. & Shah, M. Polypyrrole/MnO2 composites: synthesis, structural and electrical properties. J Mater Sci: Mater Electron 29, 9090–9098 (2018). https://doi.org/10.1007/s10854-018-8936-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-018-8936-0