
Abstract
The core–shell structure of carbon-coated MoO3 (C#MoO3) nanoparticles have been prepared by using a simple hydrothermal synthesis and using in situ method to fabricate the high performance nanocomposite with polypyrrole (PPy) and graphene nanoribbon (GNR). The chemical and structural of the samples were characterized by using Fourier transform infrared (FTIR), Raman, and X-ray diffraction. The morphology of C#MoO3 nanoparticle and its nanocomposite was also observed by using high-resolution transmission electron microscopy. The electrochemical performance of prepared PPy/C#MoO3 nanoparticle/GNR nanocomposite not only present the high specific capacitance (991 F g−1) at 5 mV s−1 scan rate in a 1 M H2SO4 electrolyte but also shows the high retention (92.1%) of capacitance after 1000 charge/discharge cycles. Electrochemical impedance spectroscopy test for PPy/C#MoO3 nanoparticle/GNR nanocomposite also shows the very low charge-transfer resistance. These superior properties significantly show that the C#MoO3 nanoparticle used to fabricate the nanocomposite can further improve the specific capacitance and cycle stability. Here this paper also provides a low cost and facile process to fabricate the high performance nanocomposite as a promising electrode material for supercapacitor.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Electrochemical capacitors, also known as supercapacitors, provide higher power density than both conventional batteries and conventional capacitors [1]. They also possess other favorable characteristics with respect to energy storage, such as having long cycling stability and possessing rapid charging and discharging rates. However, despite their excellent properties, their use in common power devices is restricted because of one specific reason: their energy density is lower than that in the batteries [2]. In order to overcome the energy density issue associated with supercapacitors, improvements will need to be made in their capacitance performance and operating voltages. Supercapacitor can be divided into two types according to their storage mechanism: electrical double-layer capacitors (EDLCs) and pseudocapacitors.
In recent years, carbon-based materials, with excellent properties (i.e., high specific surface areas, high conductivities, good structural stabilities, and environmental friendliness), such as graphene and graphene nanoribbon (GNR), have often been used as EDLC electrode materials in EDLC in supercapacitors and other micro-, nanoelectronic materials [3,4,5,6,7,8]; however, the specific capacitance of EDLCs is generally low; this results in low energy density. Therefore, many researches combine EDLC with pseudocapacitors in order to improve the low specific capacitance problem. In the last decade, EDLC and pseudocapacitor nanocomposites have been developed specifically for using it in high-performance supercapacitors [5, 9,10,11,12,13,14]. As a result, choosing good pseudocapacitor electrode materials is an important factor to consider when investigating supercapacitors. Transition metal oxides and conducting polymers are commonly used as electrode materials in pseudocapacitors; molybdenum trioxide (MoO3), in particular, has attracted a lot of attention because of its high electrochemical activity, unique structure, environmental friendliness, and cost effectiveness (its cost is lower than that of ruthenium oxide (RuO2)). However, applying MoO3 to supercapacitors is difficult because of its poor conductivity and poor cycling stability [15]. Conducting polymer polypyrrole (PPy) has also been used as a pseudocapacitor electrode material because of its high conductivity, fast redox properties, low weight, low cost and easy synthesizability [16]; however, it also has poor cycling stability due to shrinkage and swelling during charge–discharge cycles [17]. Therefore, viable methods to overcome these disadvantages of MoO3 and PPy need to be studied.
Most reports have found that combining EDLCs and pseudocapacitor nanocomposites is an effective approach in overcoming the disadvantages of these two components; for example, Xia et al. prepared a reduced-graphene oxide/MoO3/polyaniline nanocomposite with a specific capacitance of 553 F g−1 in 1 M H2SO4 at a scan rate of 1 mV s−1, and they found that it had good cycling stability after 200 cycles [18]. Das et al., meanwhile, synthesized a nanocomposite based on polyaniline, MoO3, and graphene that showed a significantly large specific capacitance of 593 F g−1; these nanocomposites presented good cycling stability even after 1000 cycles [19]. Apart from MoO3 nanocomposites, nanocomposites for other metal oxides have also been reported. Zheng et al. prepared a hierarchical reduced graphene oxide–SnO2–polypyrrole ternary composite with a specific capacitance of 280.5 F g−1; this composite showed high electrochemical performance, and it demonstrated good retention after 800 cycles [20]. Pan et al., meanwhile, prepared a new nanocomposite by combining a MnO2-coated polyaniline nanowire with a graphene sheet; this nanocomposite showed a maximum specific capacitance of 875.2 F g−1 and exhibited good cycling stability after 1000 cycles [10].
In this study, we proposed a facile and low cost method to prepare the carbon-coated MoO3 nanoparticles (C#MoO3) and its nanocomposite (PPy/C#MoO3 nanoparticle/GNR nanocomposite). By comparing this with an uncoated-MoO3 nanoparticle nanocomposite (PPy/MoO3 nanoparticle/GNR nanocomposite), we found that the electrochemical performance and cycling stability of the PPy/C#MoO3 nanoparticle/GNR nanocomposite could be increased further. The prepared C#MoO3 nanoparticles could exert three different functions: (1) promote the combination of each component through strong interactions; (2) efficiently reduce the charge-transfer resistance of the MoO3 nanoparticles by the use of the carbon-coated layer; and (3) further improve the electrochemical performance of the PPy/C#MoO3 nanoparticle/GNR nanocomposite using this carbon layer. The enhanced properties of the nanocomposite were comprehensively investigated through an electrochemical analysis. The difference in the performance between the PPy/MoO3 nanoparticle/GNR and PPy/C#MoO3 nanoparticle/GNR nanocomposites has also been investigated in this study.
2 Experimental
2.1 Preparation of GNR, MoO3 nanoparticle and C#MoO3 nanoparticle
Our carbon material of GNR was prepared by using previous research method [21]. 0.05 g multi-wall carbon nanotube (MWCNT) powders were added in 10 mL concentrated sulfuric acid (H2SO4) under stirred at room temperature for 1 h. Then 0.25 g potassium permanganate (KMnO4) was added slowly into the above solution and keeps stirred at room temperature for 1 h. After stirring, the solution was put in hot water bath (80 °C) and stirred violently for 1 h. After complete the reaction, the resulting solution was put in 5% hydrogen peroxide (H2O2) solution at room temperature for 24 h. Finally, the black powders were then filtered and washed with distilled water (DI-water), and then dried at 60 °C for 24 h.
The MoO3 nanoparticles were prepared by using simple thermal process. First, 0.137 g molybdenum chloride (MoCl5) (99.6%, Alfa Aesar) were slowly added into 10 mL methanol. Then, the solution was poured into ceramic container and dried at 70 °C. Finally, the ceramic container was put in a high temperature tube heating system and kept at 300 °C thermal treat for 1 h. After the container was cool down to room temperature, the green powders of MoO3 nanoparticle were obtained.
For the hydrothermal synthesis of core–shell structure of C#MoO3 nanoparticles, take 0.1 g MoO3 nanoparticle powders added into 20 mL DI-water and sonicated for 30 min. Then 0.2 g glucose powders were added into the suspension solution and stirred for 30 min. After stirring, the resulting solution was transferred into autoclave and hydrothermal synthesis at 180 °C for 6 h. Finally, the black solution was filtered, and washed several times with DI-water, dried at 60 °C oven for 24 h.
2.2 Preparation of PPy/C#MoO3 nanoparticle/GNR nanocomposite
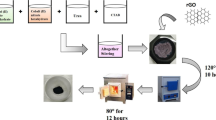
For the in situ synthesis of nanocomposite, C#MoO3 nanoparticle powders (0.008 g) and GNR (0.008 g) were dispersed simultaneously in 40 mL 1 M hydrochloric acid (HCl) solution by sonication. Then 0.08 mL pyrrole monomer was dropped in the above solution under stirred for 30 min. After that, the prepared ammonium persulfate (APS) solution (0.013 g APS powders were dissolved in 10 mL 1 M HCl) was dropped carefully into the dispersion solution at 0 °C and keeps stirred for 3 h. After reaction, the black powders were filtered and washed with DI-water and methanol successively and then dried for 1 day at 60 °C oven. The control sample (PPy/MoO3 nanoparticle/GNR nanocomposite) is also prepared by using the same method. The flow chart for prepare core–shell MoO3 nanoparticles and its nanocomposite is presented in Fig. 1.

Flow chart for the preparation of PPy/C#MoO3 nanoparticle/GNR nanocomposite
2.3 Material characterization
The Fourier transform infrared (FTIR) spectra were measured by using a Perkin–Elmer Spectrum One spectrometer with potassium bromide (KBr) pellet, and the resolution is 4 cm−1. Raman spectra were measured by using a Jobin yvon TRIAX 550 spectrometer at the scan range 500–2000 cm−1 with a wavelength 633 nm He–Ne laser. The X-ray diffraction (XRD) scan of sample was performed on a Rigaku D/MAX 2000 diffractometer equipped with Ni–filtered Cu Kα radiation in the reflection mode. The scan ranges of angle were collected from 2θ = 5°–50° with the increment of 2°/min. The morphology was observed by using a JEOL/JEM–2100F high-resolution transmission electron microscopy (HRTEM). The samples for HRTEM measurement were dispersed in ethanol. Then, the solution was dropped on lacey carbon film on copper grid.
2.4 Electrochemical performance analyses
The electrochemical performances were evaluated with a three-electrode system using 1 M H2SO4 solution as an electrolyte. 1 mg sample was dispersed into 2 mL ethanol by sonication for 1 h in presence of 0.01 mL nafion. Take 0.02 mL dispersed solution dropped onto a glassy carbon electrode with an area of 0.5 cm2 as a working electrode and dried at room temperature before test. In the conditions of electrochemical measurements, the voltage range of cyclic voltammetry (CV) and galvanostatic charge/discharge tests were measured from − 0.2 to 0.8 V (vs. Ag/AgCl). The electrochemical impedance spectroscopy (EIS) analysis was made in the frequency range from 106 to 1 Hz, using alternate current amplitude of 5 mV.
The specific capacitance (C sp) was calculated from CV curves by using the following Eq. [22]
where C sp is the specific capacitance (F g−1), \(\int {I~{\text{d}}V}\) is the integrated area of CV curve, m is the mass of sample on the working electrode, ν is the scan rate (mV s−1), V is the potential scan range.
The calculated specific capacitance from galvanostatic charge/discharge curve is using the following equation [15, 23]
where I is the discharge current (A), Δt is the discharge time (s), ΔV is the potential scan range (V), and m is the mass of sample on the working electrode (g).
3 Results and discussions
3.1 Chemical, structural and morphological characterizations
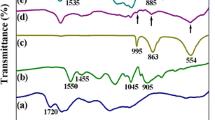
The functional groups of each material were measured using FTIR spectra, as shown in Fig. 2. In this figure, in curve a, typical PPy peaks at 1550 and 1460 cm−1, which can be attributed to the presence of C–C and C–N in-ring stretching mode, respectively. The other peaks found at 1175, 1045, and 910 cm−1 can be attributed to C–N stretching vibration, C–H deformation, and C–H out-of-plane vibration, respectively [24]. The peak at 1725 cm−1 for the GNR curve, b, was attributed to the C=O stretching vibration; it indicates the existence of oxidized functional groups in the GNR. For the pure MoO3 nanoparticles (curve c), the three main peaks appeared at 890, 830, and 775 cm−1; this was attributed to the typical characterization peaks of MoO3 monoclinic structure [25]. The FTIR spectrum of C#MoO3 nanoparticles (curve d) was obtained in order to find some information about the carbon layer on MoO3 nanoparticles. Comparing it with the pure MoO3 nanoparticle, it can be found that the appearance of the peak at 1620 cm−1 was due to the stretching mode of the C=C group, which indicates the presence of carbon. The C#MoO3 nanoparticle/GNR nanocomposite (curve e) is observed, and the appearances of the peaks of the C#MoO3 nanoparticles reveal that the C#MoO3 nanoparticles exist in the GNR matrix. Moreover, it can be observed that the peak at 1620 cm−1 (C=C group) shifted to a lower wavenumber (i.e., 1590 cm−1); this may be due to the π–π interaction between the C#MoO3 nanoparticles and the GNR. After the nanocomposite had been fabricated with PPy through in situ polymerization (curve f), the curve became similar to that of the pure PPy, and the characteristic peaks for PPy shifted to lower wavenumbers. This result was also attributed to the π–π interaction between the PPy polymer chain and the C#MoO3 nanoparticle/GNR nanocomposite [26, 27].

The FTIR spectrum of (a) Pure PPy, (b) GNR, (c) MoO3 nanoparticle, (d) C#MoO3 nanoparticle, (e) C#MoO3 nanoparticle/GNR nanocomposite, and (f) PPy/C#MoO3 nanoparticle/GNR nanocomposite
In order to provide further evidence of the existence of the carbon material, we utilized Raman spectroscope; the resulting spectra can be seen in Fig. 3. The spectra of the pure MoO3 nanoparticle are shown in curve a, and the three main peaks that appear at 901, 845, and 775 cm−1 can be attributed to the typical peaks of a MoO3 monoclinic structure; this has previously been reported [25]. For the as-prepared C#MoO3 nanoparticles (curve b), the weak peaks at 1340 and 1590 cm−1 can be attributed to the disordered (D-band) and graphitic structures (G-band), respectively. The presence of the D and G-bands further proves that the coated-carbon layer was present on the surface of the MoO3 nanoparticles.

The Raman spectrum of MoO3 nanoparticle and C#MoO3 nanoparticle
The structures of the as-prepared materials were determined using XRD technique, as shown in Fig. 4. For the pure GNR (curve a), the diffraction peak at 10.5° is due to the (001) crystal plane of the GNR layer structure. For the pure MoO3 nanoparticles (curve b), the significant diffraction peaks at 23.04°, 25.06°, and 26.08° are due to the (011), (200), and (111) crystal planes, respectively. According to the JCPDF 47–1081 database, this result proves that the MoO3 nanoparticles belong to the monoclinic (β-phase) crystal structure of MoO3. The XRD pattern of the C#MoO3 nanoparticles (curve c) shows that their diffraction peaks are the same as those of the pure MoO3 nanoparticles; this indicates that amorphous carbon coats the MoO3 nanoparticles [28]. After being combined with GNR (curve d), the C#MoO3 diffraction peaks appear in the XRD pattern of the C#MoO3 nanoparticle/GNR nanocomposite; this confirms that the C#MoO3 nanoparticles exist in the GNR matrix. For the PPy/C#MoO3 nanoparticle/GNR nanocomposite (curve e), there is appearance of C#MoO3 nanoparticle peaks, which indicates that the C#MoO3 nanoparticles are present in the nanocomposite. Furthermore, there are no new diffraction peaks in XRD profile of the PPy/C#MoO3 nanoparticle/GNR nanocomposite; this indicates that amorphous PPy coats the surface.

The XRD pattern of (a) GNR, (b) MoO3 nanoparticle, (c) C#MoO3 nanoparticle, (d) C#MoO3 nanoparticle/GNR nanocomposite, and (e) PPy/C#MoO3 nanoparticle/GNR nanocomposite
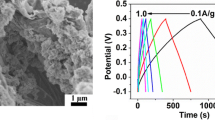
The morphologies of the MoO3 nanoparticles, C#MoO3 nanoparticles, C#MoO3 nanoparticle/GNR nanocomposite, and PPy/C#MoO3 nanoparticle/GNR nanocomposite were observed using HRTEM; they can be seen in Fig. 5. Figure 5a shows the pure MoO3 nanoparticles with diameters of approximately 20–25 nm. In Fig. 5b, the high magnification image shows the core–shell structure of the C#MoO3 nanoparticles, and the carbon layer on the nanoparticle surface can be seen to have a thickness of approximately 2 nm. In the C#MoO3 nanoparticles/GNR nanocomposite (Fig. 5c), it can be observed that the C#MoO3 nanoparticles are well loaded on the GNR surface. This result demonstrates that there is a good interaction between the C#MoO3 nanoparticles and the GNR. From the image of the PPy/C#MoO3 nanoparticle/GNR nanocomposite (Fig. 5d), it can be observed that the PPy (black arrow) coats the surface of the nanocomposite, and can also be observed that the size of the GNR nanocomposite increased to about 190 nm. Moreover, it can be seen that the C#MoO3 nanoparticles (red arrow) are present in the nanocomposite. A schematic diagram (shown in Fig. 1) of the morphologies of each sample were clearly revealed on top of the HRTEM images.

The TEM images of a MoO3 nanoparticle, b C#MoO3 nanoparticle, c C#MoO3 nanoparticle/GNR nanocomposite, and d PPy/C#MoO3 nanoparticle/GNR nanocomposite. The inset shows the schematic diagrams of morphology of each sample
3.2 Electrochemical characterizations
The electrochemical performances of the different samples were measured using cyclic voltammetry (CV), galvanostatic charge/discharge, and electrochemical impedance spectroscopy (EIS) tests. In this section, we show the difference in the electrochemical performance of the PPy/MoO3 nanoparticle/GNR and PPy/C#MoO3 nanoparticle/GNR nanocomposites. The CV curves for the PPy/MoO3 nanoparticle/GNR and PPy/C#MoO3 nanoparticle/GNR nanocomposites at a scan rate of 5 mV s−1 are shown in Fig. 6a. As demonstrated, the current density and the area of the CV curve for the PPy/C#MoO3 nanoparticle/GNR nanocomposite are larger than those of the PPy/MoO3 nanoparticle/GNR nanocomposite. In general, the specific capacitance value from the CV test depends on the area of the CV curve; therefore, this result indicates that the PPy/C#MoO3 nanoparticle/GNR nanocomposite has a better electrochemical performance and higher specific capacitance than the PPy/MoO3 nanoparticle/GNR nanocomposite. The specific capacitance (991 F g−1) of the PPy/C#MoO3 nanoparticle/GNR nanocomposite at a scan rate of 5 mV s−1 was calculated using Eq. 1. In comparison with the specific capacitance of the pure materials (i.e., pure PPy and GNR), which we investigated in our previous study, the specific capacitances of pure PPy and GNR were reported to be approximately 243 and 134 F g−1, respectively [29]. The specific capacitance of the PPy/C#MoO3 nanoparticle/GNR nanocomposite is significantly higher than that of the pure materials. Furthermore, it also shows a higher specific capacitance than that of the PPy/MoO3 nanoparticle/GNR nanocomposite (839 F g−1). The notable increase in the specific capacitance of the PPy/C#MoO3 nanoparticle/GNR nanocomposite could be attributed to the following: (1) the synergistic effect between the EDLC and the pseudocapacitor; (2) the C#MoO3 nanoparticles can efficiently reduce the charge–transfer resistance of the nanocomposite; and (3) a good combination of each component can further enhance the performance of the nanocomposite. In Fig. 6b and c, the CV curves of the PPy/MoO3 nanoparticle/GNR and PPy/C#MoO3 nanoparticle/GNR nanocomposites are shown for scan rates of 5–200 mV s−1. The closed rectangular shapes of the CV curves at various scan rates in Fig. 6c indicate that the PPy/C#MoO3 nanoparticle/GNR nanocomposite possesses a better charge propagation property than the PPy/MoO3 nanoparticle/GNR nanocomposite [30, 31]. The trends of the calculated specific capacitances at various scan rates are shown in Fig. 6d. We found that the specific capacitance of all of the nanocomposites decreased when the scan rate was increased; this is because it is not possible for the charge to completely diffuse into inner active materials at high scan rates, and this results in a relatively lower specific capacitance [32].

a CV curves of PPy/MoO3 nanoparticle/GNR nanocomposite and PPy/C#MoO3 nanoparticle/GNR nanocomposite at a scan rate of 5 mV s−1, CV curves of b PPy/MoO3 nanoparticle/GNR nanocomposite and c PPy/C#MoO3 nanoparticle/GNR nanocomposite at various scan rates, and d The calculated specific capacitance at various scan rates
The electrochemical performance of the nanocomposites was measured using a galvanostatic charge/discharge test at a current density of 5 A g−1. Figure 7a shows the curve obtained for the PPy/C#MoO3 nanoparticle/GNR nanocomposite; it is almost triangular in shape, which indicates that the PPy/C#MoO3 nanoparticle/GNR nanocomposite possesses a good charge propagation property. A superior specific capacitance of 960 F g−1 at a current density of 5 A g−1 was calculated for the PPy/C#MoO3 nanoparticle/GNR nanocomposite using Eq. 2; this value was greater than that for the PPy/MoO3 nanoparticle/GNR nanocomposite (816 F g−1); this result is similar to that found in the CV test. The long cycling stability of the two nanocomposites was evaluated using charge/discharge tests at a current density of 15 A g−1 for 1000 cycles, as shown in Fig. 7b. In the inset of Fig. 7b, we can see that the reduced discharge time of the PPy/MoO3 nanoparticle/GNR nanocomposite is larger than that of the PPy/C#MoO3 nanoparticle/GNR nanocomposite after 1000 cycles. The retention of the specific capacitance calculated for the PPy/C#MoO3 nanoparticle/GNR nanocomposite is approximately 92.1%, which is better than the 81.6% found for the PPy/MoO3 nanoparticle/GNR nanocomposite. The good cycling stability of the PPy/C#MoO3 nanoparticle/GNR nanocomposite may be due to the carbon layer that coats the surface of the MoO3 nanoparticle surface and that subsequently improves the stability of the MoO3 nanoparticles and nanocomposite. In general, because of the advantages offered by EDLCs, the carbon layer and the GNR serve as an EDLC in the nanocomposite and provide it with good cycling stability required for overcoming the poor cycling stability of the pseudocapacitor. Therefore, the enhancement in the cycling stability of the PPy/C#MoO3 nanoparticle/GNR nanocomposite can be attributed to the synergistic effect and the strong interaction between the EDLC and the pseudocapacitor [33, 34].

a Galvanostatic charge/discharge curves of PPy/MoO3 nanoparticle/GNR nanocomposite and PPy/C#MoO3 nanoparticle/GNR nanocomposite at a current density of 5 A g−1, and b Cycle stability of PPy/MoO3 nanoparticle/GNR nanocomposite and PPy/C#MoO3 nanoparticle/GNR nanocomposite at a current density 15 A g−1. The inset shows the discharge curves of the first and last cycle for the PPy/MoO3 nanoparticle/GNR nanocomposite and PPy/C#MoO3 nanoparticle/GNR nanocomposite
EIS analysis is a powerful tool for investigating the impedance behavior of electrode materials. The typical Nyquist plots of the MoO3 nanoparticles, C#MoO3 nanoparticles, PPy/MoO3 nanoparticle/GNR nanocomposite, and PPy/C#MoO3 nanoparticle/GNR nanocomposite are shown in Fig. 8. Generally, in the high-frequency region, the solution resistance (R s) is calculated from the intercept with the real axis [35]. The diameter of the semi-circle in the high-frequency region is attributed to the charge-transfer resistance (R ct) at the electrode–electrolyte interface [36]. The calculated R s and R ct values of all of the samples are shown in Table 1. According to previous studies, low R ct values indicate that there is a low charge-transfer resistance at the electrode–electrolyte interface; this could improve the electrochemical performance [37, 38]. From Table 1, it can be seen that the R ct values of the C#MoO3 nanoparticles and PPy/C#MoO3 nanoparticle/GNR nanocomposite are clearly lower than those of the pure MoO3 nanoparticles and PPy/MoO3 nanoparticle/GNR nanocomposite; this indicates that the charge-transfer resistance of the C#MoO3 nanoparticles and PPy/C#MoO3 nanoparticle/GNR nanocomposite are low in value. This result therefore proves that the C#MoO3 nanoparticles used to make the PPy/C#MoO3 nanoparticle/GNR nanocomposite can efficiently reduce the charge-transfer resistance and improve the electrochemical performance of the nanocomposite. Furthermore, in the low-frequency region, the portion of the Nyquist plot with a 45° slope represents the resistance to ion diffusion in the electrode materials [15]. The slopes of the C#MoO3 nanoparticles and PPy/C#MoO3 nanoparticle/GNR nanocomposite are larger than those of the MoO3 nanoparticles and PPy/MoO3 nanoparticle/GNR nanocomposite; this indicates that the ion diffusion resistance for the C#MoO3 nanoparticles and PPy/C#MoO3 nanoparticle/GNR nanocomposite is low; as a result, charge can easily diffuse into the inner spaces of the electrode materials.

The Nyquist plots of a MoO3 nanoparticle and C#MoO3 nanoparticle, and b PPy/MoO3 nanoparticle/GNR nanocomposite and PPy/C#MoO3 nanoparticle/GNR nanocomposite
4 Conclusions
In conclusion, we used a core–shell structure of C#MoO3 nanoparticles to fabricate a high-performance PPy/C#MoO3 nanoparticle/GNR nanocomposite via an in situ chemical oxidation polymerization. The presence of amorphous carbon layer on the surfaces of the MoO3 nanoparticles was successfully confirmed using Raman spectroscopy and XRD measurements. The FTIR spectra showed that the π–π interaction between PPy, the C#MoO3 nanoparticles, and the GNRs improved the combinations between these components; this was also indicated by the HRTEM images. The CV test and galvanostatic charge/discharge test show that the PPy/C#MoO3 nanoparticle/GNR nanocomposite exhibited the highest specific capacitance of about 991 and 960 F g−1 in the respective tests. The PPy/C#MoO3 nanoparticle/GNR nanocomposite was also found to have excellent cycling stability (92.1% retention) after 1000 charge/discharge cycles; this was about 10% greater than that of the PPy/MoO3 nanoparticle/GNR nanocomposite. Furthermore, the EIS results show that the charge-transfer resistance values of the C#MoO3 nanoparticles and PPy/C#MoO3 nanoparticle/GNR nanocomposite were significantly lower than those of the MoO3 nanoparticles and PPy/MoO3 nanoparticle/GNR nanocomposite. These results indicate that the carbon layer, which coats the MoO3 nanoparticle surface, can improve the performance of the nanocomposite. Owing to the superior electrochemical performance found in this study, we believe that the PPy/C#MoO3 nanoparticle/GNR nanocomposite will be a promising supercapacitor electrode material.
References
P. Simon, Y. Gogotsi, Materials for electrochemical capacitors. Nat. Mat. 7, 845–854 (2008)
G.X. Pan, X.H. Xia, F. Cao, J. Chen, P.S. Tang, Y.J. Zhang, H.F. Chen, High-performance asymmetric supercapacitors based on core/shell cobalt oxide/carbon nanowire arrays with enhanced electrochemical energy storage. Electrochim. Acta 133, 522–528 (2014)
E. Frakowiak, F. Béguin, Carbon materials for the electrochemical storage of energy in capacitors. Carbon 39, 937–950 (2001)
K.I. Bolotin, K.J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim, H.L. Stormer, Ultrahigh electron mobility in suspended graphene. Solid State Commun. 146, 351–355 (2008)
H.P. Oliveira, S.A. Sydlik, T.M. Swager, Supercapacitors from free-standing polypyrrole/graphene nanocomposites. J. Phy. Chem. C 117, 10270–10276 (2013)
L. Li, A.R.O. Raji, H.L. Fei, Y. Yang, ELG Samuel, J.M. Tour, Nanocomposite of polyaniline nanorods grown on graphene nanoribbons for highly capacitive pseudocapacitors. ACS Appl Mat Interfaces 5, 6622–6627 (2013)
M.K. Liu, W.W. Tjiu, J.S. Pan, C. Zhang, W. Gao, T.X. Liu, One-step synthesis of graphene nanoribbon-MnO2 hybrids and their all-solid-state asymmetric supercapacitors. Nanoscale 6, 4233–4242 (2014)
F. Akbar, M. Kolahdouz, S. Larimian, B. Radfar, H.H. Radamson, Graphene synthesis, characterization and its applications in nanophotonics, nanoelectronics, and nanosensing. J. Mater. Sci. 26, 4347–4379 (2015)
F. Yang, L.B. Zhang, A. Zuzuarregui, K. Gregorczyk, L. Li, M. Beltràn, C. Tollan, J. Brede, C. Rogero, A. Chuvilin, M. Knez, Functionalization of defect sites in graphene with RuO2 for high capacitive performance. ACS Appl. Mat. Interfaces 7, 20513–20519 (2015)
C. Pan, H.T. Gu, L. Dong, Synthesis and electrochemical performance of polyaniline @MnO2/graphene ternary composites for electrochemical supercapacitors. J. Power Sources 303, 175–181 (2016)
J. Zhang, X.S. Zhao, Conducting polymers directly coated on reduced graphene oxide sheets as high-performance supercapacitor electrodes. J. Phy. Chem. C 116, 5420–5426 (2012)
Y. Li, G. Louarn, P.H. Aubert, V. Alain-Rizzo, L. Galmiche, P. Audebert, F. Miomandre, Polypyrrole-modified graphene sheet nanocomposites as new efficient materials for supercapacitors. Carbon 105, 510–520 (2016)
M. Manoj, K.M. Anilkumar, B. Jinisha, Jayalekshmi, Polyaniline-graphene oxide based ordered nanocomposite electrodes for high-performance supercapacitor application. J. Mater. Sci. 28, 14323–14330 (2017)
R. Dhilip Kumar, Y. Andou, Karuppuchamy, Facile synthesis of Co–WO3/functionalized carbon nanotube nanocomposites for supercapacitor applications. J. Mater. Sci. 28, 5425–5434 (2017)
X. Zhang, X.Z. Zeng, M. Yang, Y.X. Qi, Investigation of a branchlike MoO3/polypyrrole hybrid with enhanced electrochemical performance used as an electrode in supercapacitors. ACS Appl. Mat. Interfaces 6, 1125–1130 (2014)
T.M. Wu, S.H. Lin, Synthesis, characterization, and electrical properties of polypyrrole/multiwalled carbon nanotube composites. J. Polym. Sci. A 44, 6449–6457 (2006)
J. Zhang, L.B. Kong, J.J. Cai, Y.C. Luo, L. Kang, Nano-composite of polypyrrole/modified mesoporous carbon for electrochemical capacitor application. Eletrochim Acta 55, 8067–8073 (2010)
X.F. Xia, Q.L. Hao, W. Lei, W.J. Wang, H.L. Wang, X. Wang, Reduced-graphene oxide/molybdenum oxide/polyaniline ternary composite for high energy density supercapacitors: synthesis and properties. J. Mater. Chem. 22, 8314–8320 (2012)
A.K. Das, S.K. Karan, B.B. Khatua, High energy density ternary composite electrode material based on polyaniline (PANI), molybdenum trioxide (MoO3) and graphene nanoplatelets (GNP) prepared by sono-chemical method and their synergistic contributions in superior supercapacitive performance. Electrochim. Acta 180, 1–15 (2015)
W.Q. Zheng, S.B. Li, X.H. Yu, C.L. Chen, H.B. Huang, Y.N. Huang, L. Li, Synthesis of hierarchical reduced graphene oxide–SnO2–polypyrrole ternary composites with high electrochemical performance. Mat. Res. Bull. 80, 303–308 (2016)
D.V. Kosynkin, A.L. Higginbotham, A. Sinitskii, J.R. Lomeda, A. Dimiev, D.K. Price, J.M. Tour, Longitudinal unzipping of carbon nanotubes to form graphene nanoribbons. Nature 458, 872–877 (2009)
A. Davies, P. Audette, B. Farrow, F. Hassan, Z.W. Chen, J.Y. Choi, A.P. Yu, Graphene-based flexible supercapacitors: pulse-electropolymerization of polypyrrole on free-standing graphene films. J. Phys. Chem. C 115, 17612–17620 (2011)
Y.Q. Zhao, M. Lu, P.Y. Tao, Y.J. Zhang, X.T. Gong, Z. Yang, G.Q. Zhang, H.L. Li, Hierarchically porous and heteroatom doped carbon derived from tobacco rods for supercapacitors. J. Power Sources 307, 391–400 (2016)
X.J. Lu, F. Zhang, H. Dou, C.Z. Yuan, S.D. Yang, L. Hao, L.F. Shen, L.J. Zhang, X.G. Zhang, Preparation and electrochemical capacitance of hierarchical graphene/polypyrrole/carbon nanotube ternary composites. Electrochim. Acta 69, 160–166 (2012)
P. Thi Thuy Phuong, N. Phuc Hoang Duy, V. Tan Tai, N. Huu Huy Phuc, L. Cam Loc, Facile method for synthesis of nanosized β-MoO3 and their catalytic behavior for selective oxidation of methanol to formaldehyde. Adv. Nat. Sci. 6, 045010 (2015)
H. Wang, Q. Hao, X. Yang, L. Lu, X. Wang, Graphene oxide doped polyaniline for supercapacitors. Electrochem. Commun. 11, 1158–1161 (2009)
R. Bissessur, K.Y. Peter Liu, S.F. Scully, Intercalation of polypyrrole into graphite oxide. Synth. Met. 156, 1023–1027 (2006)
M.F. Hassan, Z.P. Guo, Z. Chen, H.K. Liu, Carbon-coated MoO3 nanobelts as anode materials for lithium-ion batteries. J. Power Sources 195, 2372–2376 (2010)
F.H. Hsu, T.M. Wu, Poypyrrole/molybdenum trioxide/graphene nanoribbon ternary nanocomposite with enhanced capacitive performance as an electrode for supercapacitor. J. Solid State Electrochem. 20, 691–698 (2016)
P. Si, S.J. Ding, X.W. Lou, D.H. Kim, An electrochemically formed three-dimensional structure of polypyrrole/graphene nanoplatelets for high-performance supercapacitors. RSC Adv. 1, 1271–1278 (2011)
H. Chen, M.B. Müller, K.J. Gilmore, G.G. Wallace, D. Li, Mechanically strong, electrically conductive, and biocompatible graphene paper. Adv. Mater. 20, 3557–3561 (2008)
W.J. Wang, Q.L. Hao, W. Lei, X.F. Xia, X. Wang, Graphene/SnO2/polypyrrole ternary nanocomposites as supercapacitor electrode materials. RSC Adv. 2, 10268–10274 (2012)
J.H. Liu, J.W. An, Y.X. Ma, M.L. Li, R.B. Ma, Synthesis of a graphene-polypyrrole nanotube composite and its application in supercapacitor electrode batteries and energy storage. J. Electrochem. Soc. 159, A828–A833 (2012)
L. Li, H. Song, Q. Zhang, J. Yao, X. Chen, Effect of compounding process on the structure and electrochemical properties of ordered mesoporous carbon/polyaniline composites as electrodes for supercapacitors. J. Power Sources 187, 268–274 (2009)
Y. Liu, B.H. Zhang, Y.Q. Yang, Z. Chang, Z.B. Wen, Y.P. Wu, Polypyrrole-coated α-MoO3 nanobelts with good electrochemical performance as anode materials for aqueous supercapacitors. J. Mater. Chem. A 1, 13582–13587 (2013)
S. Sahoo, S. Dhibar, G. Hatui, P. Bhattacharya, C.K. Das, Graphene-polypyrrole nanofiber nanocomposite as electrode material for electrochemical supercapacitor. Polymer 54, 1033–1042 (2013)
X. Wang, C. Yang, H.D. Li, P. Liu, Synthesis and electrochemical performance of well-defined flake-shaped sulfonated graphene/polypyrrole composites via facile in situ doping polymerization. Electrochim. Acta 111, 729–737 (2013)
J. Wang, Y. Li, J. Ge, B.P. Zhang, W. Wan, Improving photocatalytic performance of ZnO via synergistic effects of Ag nanoparticles and graphene quantum dots. Phys. Chem. Chem. Phys. 17, 18645–18652 (2015)
Acknowledgements
The financial support provided by Ministry of Science and Technology through the project MOST 104-2212-E-005-089-MY2 is greatly appreciated.
Funding
Funding was provided by Ministry of Science and Technology, Taiwan (Grand No. MOST 104-2212-E-005-089-MY2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hsu, FH., Wu, TM. Facile synthesis of polypyrrole/carbon-coated MoO3 nanoparticle/graphene nanoribbon nanocomposite with high-capacitance applied in supercapacitor electrode. J Mater Sci: Mater Electron 29, 382–391 (2018). https://doi.org/10.1007/s10854-017-7927-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-7927-x