
Abstract
The solid solution of multiferroic BiFeO3 with antiferroelectric NaNbO3 [i.e., Bi1−x Na x Fe1−x Nb x O3] for x = 0.20, 0.25, 0.30, 0.32, 0.38 and 0.40 prepared by means of high temperature solid state ceramic method are presented. X-ray diffraction analysis confirms the single-phase formation. Structural transformation in solid solution from rhombohedral to cubic phase has been observed with increasing NaNbO3 concentration. The field emission scanning electron microscopy shows the formation of dense ceramic with a non-uniform grain size. Room temperature Raman studies also confirms a structural transformation in solid solution with increase in NaNbO3 concentration. The structural transformation in solid solution has been explained on the basis of lattice strain induced by doping.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Multiferroics have attracted many researchers due their potential applications such as electrical transformers, sensors, integrated circuits and storage devices [1]. There are very few multiferroics exist in the nature or can be synthesized in laboratory because of various factors such as difficulties in coupling between spin and charge.
Among the various multiferroics, Bismuth ferrite (BiFeO3), known to be a single phase material with distorted perovskite (ABO3) structure, is one of the most extensively studied room temperature (RT) multiferroic [2]. BiFeO3 has exceptional applications such as photo catalytic, ultrafast, optoelectronic and infrared detector [3, 4]. The BiFeO3 is unique, as its ferroelectric and magnetic transition temperatures are well above the RT (i.e. T C~1103 K and T N~643 K respectively) which opens the possibility of potential devices based on magneto-electric (ME) coupling operating above RT [5, 6]. In BiFeO3, the G-type antiferromagnetic structure is modulated to a spiral spin structure, whose modulation vector has a long period of λ = 620–640 Å [7, 8]. In spite of various peculiar qualities, BiFeO3 has some inherent problems that limit its utilization for device applications. For instance, BiFeO3 has high leakage current which allow passing current through it when a high voltage is applied. It also has high dielectric loss and low polarization [9]. In order to solve these problems, several attempts have been made such as doping at the appropriate atomic sites and/or fabricating its composites and solid solutions with other perovskites.
The anti-ferroelectric NaNbO3 is one of the representative lead-free piezoelectric perovskite oxides which have large practical importance. The high dielectric constant and low loss of NaNbO3 is supposed to improve the electrical and dielectric properties of BiFeO3. Several research groups have reported the solid solution of BiFeO3 with NaNbO3 and the structural, magnetic and ferroelectric phase transitions have been investigated [10]. Therefore, solid-solution of BiFeO3 with NaNbO3 is of considerable interest.
The majority of the reports for solid-solution of BiFeO3 with NaNbO3 are focused on the effects of doping on multiferroic properties, while structural evolution with composition has received comparatively less attention. In PbTiO3–PbZrO3 system, enhancements in electro-mechanical properties are found at morphotropic phase boundary (MPB) [11]. Raevski et al. [12] found that the solid solutions of (1−x)BiFeO3–xNaNbO3 exhibit rhombohedral for x ≤ 0.25 and orthorhombic distortion of perovskite structure for x ≥ 0.90 while compositions ranging from 0.25 ≤ x ≤ 0.90 stabilize in cubic structure. In Bi1− x Na x Fe1−x Nb x O3 system, when x = 0.1, the sample retains rhombohedral perovskite structure but for x = 0.3 and x = 0.5 samples crystallize pseudo-cubic or cubic structure showing structural transition, as is reported by Ma et al. [13]. Whereas, Dash et al. [14] reports the tetragonal phase for x = 0.2 and 0.3 and enhancement of spontaneous polarization with increase of x. Recently, Ummer et al. have reported the high dielectric constant, good ME coupling and improved optical properties of BiFeO3–NaNbO3 nano-composite sample [15]. Therefore, it is of great importance to identify the composition range in which structural transformation occurs and find out the MPB in BiFeO3–NaNbO3 solid solution. In present paper, solid solutions of BiFeO3 and NaNbO3 in different compositional range with a general formula Bi1−x Na x Fe1−x Nb x O3 where x = 0.20, 0.25, 0.30, 0.32, 0.38, and 0.40, using a high temperature solid-state reaction method are presented. The primary aim of the present work is to address structural transformation in BiFeO3–NaNbO3 solid solution. The structural characterizations of these solid solutions have been carried out by using X-ray diffraction (XRD) and micro-Raman spectroscopy. Micro-structural and elemental analyses are carried out by field emission scanning electron microscope (FESEM) and energy dispersive X-ray analysis (EDX), respectively.
2 Experimental
The polycrystalline samples of Bi1−x Na x Fe1−x Nb x O3 with x = 0.20, 0.25, 0.30, 0.32, 0.38 and 0.40 were prepared by solid state ceramic method from high-purity ingredients (without further purification) such as, Bi2O3 (99% pure, M/s LOBA Chemie Pvt. Ltd., India), Fe2O3 (98.5% purity, M/s LOBA Chemie Pvt. Ltd., India), Nb2O5 (99.9%, M/s Himedia Chemie Pvt. Ltd., India), and Na2CO3 (99%, M/s MERCK Chemie Pvt. Ltd., India). The above ingredients in suitable stoichiometry were mixed thoroughly with acetone medium by mortar-pestle in air for 4 h and then calcined at 750 °C for 4 h. The calcined powders were then cold pressed using a hydraulic press by applying pressure of 4 × 106 Nm− 2 into pellets of 10 mm diameter and thickness of 2 mm. These pellets were then sintered at 870 °C for 8 h.
The structural analysis of the calcined powders as well as sintered samples were carried out using X-ray powder diffractometer (Rigaku SmartLab, Japan) with CuKα radiation (λ =1.54056 Å). The microstructures of the samples were recorded using a scanning electron microscope (Carl Zeiss, Japan) operated at 20 kV. The Raman spectra of sintered samples were obtained by micro-Raman spectrometer (STR-500, Japan) at RT.
3 Results and discussion
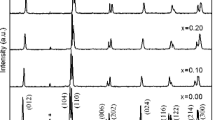
Figure 1 shows the XRD pattern of Bi1− x Na x Fe1−x Nb x O3 for x = 0.20, 0.25, 0.30, 0.32, 0.38 and 0.40 samples which clearly evidenced the formation of single phase materials. All samples have a major phase with perovskite structure; however a trace amount of secondary phase of Bi24FeO39 and Bi2Fe4O9 are also detected, as is marked with * and # in the diffraction pattern, respectively. The diffraction peaks of the samples have been indexed using POWDMULT computer software [16]. Figure 2a–c shows the extended region of Fig. 1 for different 2θ values of 31–33°, 37.5–41.5°and 54–60° to clearly evidence the diffraction peak positions. The two diffraction peak at 31.82° and 32.11° corresponding to the (104) and (110) lattice plane of rhombohedral phase are assigned for samples x = 0.20, 0.25 and 0.30, marked as R in the diffraction pattern. These doubly split peaks merge to form a broadened diffraction peak positioned at 31.98° with the increase in NaNbO3 concentration for x = 0.32 which can be assign to the (110) lattice plane of cubic phase, marked C in the pattern. Further, the increase of NaNbO3 concentration from x = 0.32 to x = 0.38 and x = 0.40, the peak is shifted at 32.0°. The similar trend of shifting of diffraction peaks are also observed for the 2θ values of 37.5–41.5° and 54–60°, as is shown in Fig. 2b, c. These results indicate that the solid-solutions of Bi1− x Na x Fe1−x Nb x O3 systems change from rhombohedral to cubic structure for x = 0.32 composition. Therefore, MPB for presently studied materials Bi1− x Na x Fe1−x Nb x O3 is at for x = 0.31 i.e. below x = 0.31 the prepared materials Bi1− x Na x Fe1−x Nb x O3 are in rhombohedral structure and above x = 0.31 the prepared materials Bi1− x Na x Fe1−x Nb x O3 are in cubic structure. A noticeable width in diffraction peaks is also observed which shows the nano-crystalline nature of solid solution. The number of possible parameter including micro-strain, crystal size effect, grain surface relaxation and formation of point defects contributes to the broadening of diffractions peaks [17–19]. The average crystallites sizes (D) of all prepared samples are given in Table 1, as estimated by Debye-Scherer’s formula [20, 21]. The Debye-Scherer’s formula provides the correct crystallite size values only when the diffracting material is stress free [22]. However, in present studies, samples are doped with Na and Nb at different lattice sites. Therefore, the presence lattice strain is expected in the samples. The non-uniform strain in unit cells gives peak broadening in diffraction patterns. In order to have some understanding of stress, Williamson-Hall plot have been drawn for all samples [23]. The strain is estimated from the slope of the fitting straight line by the following equation,

XRD pattern of Bi1− x Na x Fe1−x Nb x O3 samples for x = 0.20, 0.25, 0.30, 0.32, 0.38 and 0.40 sintered at 870 °C for 8 h. All samples show single phase formation with perovskite structure. The R represents the rhombohedral phase and C represents the cubic phase

The extended region of Fig. 1 for different 2θ values of a 31–33°, b 37.5–41.5°and c 54–60° to clearly evidence the diffraction peak shift. The R represents the rhombohedral phase and C represents the cubic phase. The MPB is at x = 0.31, below which Bi1− x Na x Fe1−x Nb x O3 are in rhombohedral structure and above x = 0.31 the prepared materials Bi1− x Na x Fe1−x Nb x O3 are in cubic structure
where, ε is the strain present in sample, β is the full width at half maxima (FWHM) of the diffraction peak, λ is the wavelength of X-ray, θ is the Bragg’s angle, D is crystallite/grain size and k is the Debye–Scherrer constant (shape factor). Figure 3 shows Williamson-Hall plots for all samples. The slope of the fitted straight line increases with increasing NaNbO3 concentration (x) which clearly depict that the significant amount of strain is present in the samples and is larger for higher doping concentration. The increase of strain in the solid solution can be understood on the basis of valance state of host and ionic radius of substituted ions. In present study, we expected that Na+ is substituted at Bi3+ site and Nb5+ is substituted at Fe3+ site. The ionic radius of Bi3+, Na+, Fe3+ and Nb5+ are 1.82, 1.86, 1.26 and 1.46 Å, respectively. Since the ionic radius of Nb is much greater that of Fe, therefore the substitution of more Nb at Fe site may distort in FeO6 octahedra [24] The lattice site and valance state mismatch produces site disorder, and vacancies which lead to the increase of lattice strain in the samples. The increase of strain in the samples possibly shifts the diffractions peak position and the possible structural phase of the material. Zhai et al. [25] have observed similar strain-induced structural phase transition in Bi1−x Tb x FeO3 thin films.

Williamson-Hall Plot for Bi1− x Na x Fe1−x Nb x O3; a x = 0.25, b x = 0.32, c x = 0.38 and d x = 0.40 compositions. The positive slope of the linear fit indicates the presence of tensile stress. The slope in the samples increases with increasing doping concentration indicates the increase of strain with doping concentration
The microstructures analyses of the samples have been investigated by means of FE-SEM measurements. Figure 4 shows the FE-SEM images of Bi1− x Na x Fe1−x Nb x O3 for x = 0.32, 0.38 and 0.40. The SEM micrographs depict a few pores at the grain boundaries; however cracks or micro cracks are not detected in the samples. The grains are nearly spherical in shape and closely packed indicating the formation of highly dense samples. It is also observed that the shape and size of the grains are not uniform. Energy dispersive X-ray analysis (EDX) is an analytical technique used for the elemental analysis or chemical characterization of a sample. The corresponding EDX spectra the samples are shown in insets of Fig. 4. No extra peaks have been traced out which confirms that there is no contamination in the samples. The elemental ratios of the samples are close to the empirical formula. In order to have deep insight of elemental distribution among grain and grain boundaries, regional element mapping have been performed, as depicted in Fig. 5. One can see that all of the dopants are homogeneously distributed throughout the samples, regardless of the grain size. The elements, especially Bi, Fe, Na, Nb and O were observed to be homogeneously distributed across the grains and grain boundaries.

SEM and EDXA (inset) image for the samples Bi1− x Na x Fe1−x Nb x O3; a x = 0.32, b x = 0.38 and c x = 0.40. The grains are nearly spherical in shape and size of the grains is not uniform

Elemental mapping of the samples for composition x = 0.20 at a marked selected region. All the constituent elements are present over the all surface. There is no contamination present in the samples
Figure 6 shows the Raman spectra of Bi1− x Na x Fe1−x Nb x O3 for x = 0.20, 0.25, 0.32, 0.38 and 0.40. The peaks at 153, 170 and 236 cm− 1 could be assigned as A1-1, A1-2 and A1-3 phonon modes of BiFeO3, respectively. Other phonon modes are located in the range of 250–850 cm− 1. The Raman peaks below 250 cm− 1, assigned to A1 modes, are related to Bi–O vibrations [26]. Theoretical analysis has predicted that there are 18 optical phonon modes: 4A1 + 5A2 + 9E for BiFeO3 at room temperature. The A1 and E modes are both Raman and IR-active modes, whereas the A2 modes are Raman and IR inactive modes [27]. It is observed that A1-1 and A1-2 modes shift to higher frequency site and broaden gradually with increase in the substitution concentration. The shift of A1-1 and A1-2 phonon modes can be understood on the basis of model proposed by Do et al. [28]. If k is the force constant and M is reduced mass then the frequency of the mode is proportional to (k/M)½. Since the atomic mass of Na is less than that of Bi which causes the shifting of A1-1 and A1-2 phonon modes to higher frequency side. This phenomenon is due to the chemical pressure-induced bond shortening and lattice distortion. The decrease in intensity of A1-1 and A1-2 mode in the substituted BFO indicates the decline of stereo-chemical activity of Bi3+ lone pair electrons which in turns changes the Bi–O bonds [29]. Raman modes above 200 cm− 1 are related to stretching and bending mode for FeO6 octahedra [30]. Raman mode near at 483 cm− 1 attributed to change in displacement in B-site cations and octahedral tilt. Under high lattice strain, the volume of the unit cell is compressed, consequently leading to the reduction of the octahedral tilting.

Room temperature RAMAN spectra for Bi1− x Na x Fe1−x Nb x O3; x = 0.20, 0.25, 0.32, 0.38 and 0.40. The dotted line clearly shows shift in Raman peak due to increase of doping concentration which also confirms the structural phase transformation
The origin of this octahedral tilting and phase transition can also be understood on the basis of Goldschmidt’s tolerance factor (t) which associated with cationic size mismatch on both site. The tolerance factor can be given by,
where R A, R B and R O are the average radii A-site, B-site and ionic radius of O2−; t is unity for perfect perovskite structure (ABO3) i.e. for undistorted structure; when t < 1 then distortion occur in the perovskite structure indicating tilting or rotation in FeO6 octahedra. We have calculated tolerance factor using Eq. (2) and is found to be 0.8904, 0.8871, 0.8823, 0.8784 and 0.8770 for x = 0.20, 0.25, 0.32, 0.38 and 0.40, respectively. The decrease of tolerance factor with doping concentration confirms the increase of octahedral rotation. It is well established that if the tolerance factor is less than unity, oxygen octahedra tends to rotate to realize the lattice [31]. In present study, the continuous increase of oxygen octahedra rotation makes rhombohedral phase unstable and cubic phase stable at higher doping concentration. Therefore, we observed peaks shifting in diffraction pattern.
4 Conclusions
We have synthesized Bi1− x Na x Fe1−x Nb x O3 system for x = 0.20, 0.25, 0.30, 0.32, 0.38 and 0.40 using a conventional solid state method. All compositions have a major phase with perovskite structure. A trace amount of secondary phases (Bi2Fe4O9 and Bi24FeO39) are also detected. A structural transformation from rhombohedral to cubic phase is observed with doping concentration. The MPB for Bi1− x Na x Fe1−x Nb x O3 is observed at x = 0.31 i.e. below x = 0.31 the prepared materials are in rhombohedral structure and above x = 0.31 are in cubic structure. The micro-structural analyses of the samples show that the grains are in spherical shape but not uniform. The Raman peaks were found to be sifted due to strain induced by the doping. The structural phase transitions have been explained on the basis of lattice strain and octahedra tilting occurring in unit cell.
References
J. Wang, J.B. Neaon, H. Zheng, V. Nagarjan, S.B. Ogale, B. Liu, D. Viehland, V. Vaithyanan, D.G. Schlom, U.V. Waghmae, N.A. Spaldin, K.M. Rabe, M. Wuttig, R. Ramesh, Science. 299, 1719–1722, (2003)
V.F. Freitas, O.A. Protzek, L.A. Montoro, A.M. Gonçalves, D. Garcia, J.A. Eiras, R. Guo, A.S. Bhalla, L.F. Coticaad, I.A. Santos, J. Mater. Chem. C. 2, 364–372 (2014)
S.R. Basu, L.W. Martin, Y.H. Chu, M. Gajek, R. Ramesh, R.C. Rai, X. Xu, J.L. Musfeld, Appl. Phys. Lett. 92, 091905, (2008)
A.J. Hauser, J. Zhang, L. Mier, R.A. Ricciardo, P.M. Woodward, T.L. Gustafson, L.J. Brillson, F.Y. Yang, Appl. Phys. Lett. 92, 2901 (2008)
N.A. Hill, J. Phys. Chem. B. 104, 6694–6709, (2000)
M. Amin, H.M. Rafique, M. Yousaf, S.M. Ramay, S. Atiq, J. Mater. Sci. 27, 11003–11011, (2016)
D.I. Khomskii, Physics. 2, 20, (2009)
Q.X. Xing, Z. Han, S. Zhao, J. Mater. Sci. DOI:10.1007/s10854-016-5524-z, (2016)
Y. Wang, C.W. Nan, Appl. Phys. Lett. 89, 052903–052905 (2006)
G. Li, Z. Yi, Y. Bai, W. Zhang, H. Zhang, Dalton Trans. 41,110194–10198, (2012)
B. Jaffe, C.R. Cook, H. Jaffe, Piezo-electric ceramics. (Academic Press, London, 1971)
P. Raevski, S.P. Kubrin, J.L. Dellis, S.I. Raevskaya, D.A. Sarychev, V.G. Smotrakov, V.V. Eremkin, M.A. Seredkina, Ferroelectrics, 371, 113–118, (2008)
Y. Ma, X.M. Chen, J. Appl. Phys. 105, 054107 (2009)
S. Dash, R. Padhee, P.R. Das, R.N.P. Choudhary, J. Mater. Sci. 24, 3315–3323 (2013)
R. P. Ummer, P. Sreekanth, B. Raneesh, R. Philip, D. Rouxel, S. Thomas, N. Kalarikkal, RSC Adv. 5, 67157–67164, (2015)
E.W. Powd An interactive powder diffraction data interpretation and indexing program, Version 2.1 (Flinders University of South Australia, Bedford Park, 1989)
S. Rana, S. Ram, S. Seal, S.K. Roy, Appl. Surf. Sci. 236, 141–154 (2004)
R. Elilarassi, G. Chandrasekaran, J. Mater. Sci. Mater. Electron. 22, 751–756, (2011)
S.M. Salaken, E. Farzan, J. Podder, J. Semicond. 34, 073003, (2013)
H.P. Klung, L.B. Alexander, X-ray diffraction procedures (Wiley, New York, 1974), p. 687
B.D. Cullity, Elements of X-ray diffraction (Addison-Wesley Publishing Company, Inc., Philippines, 1978), p. 284
X.M. Duan, C. Stampfl, M.M.M. Bilek, D.R. McKenzie, Phys. Rev. B. 79, 235208 (2009)
G.K. Williamson, W.H. Hall, Acta Metall. 1, 22–31 (1953)
E. Chandrakala, J.P. Praveen, A. Kumar, A.R. James, D. Das, J. Am. Ceram. Soc. (2016). doi:10.1111/jace.14409
X. Zhai, H. Deng, W. Zhou, P. Yang, J. Chu, J. Phys. D. 48, 385002, (2015)
H. Fukumura, H. Harima, K. Kisoda, M. Tamada, Y. Noguchi, M. Miyayama, J. Magn. Magn. Mater. 310, 367–369, (2007)
D. Kothari, V. R. Reddy, V. G. Sathe, A. Gupta, A. Banerjee, A. M. Awasthi, J. Magn. Magn. Mater. 320, 548–552, (2008)
D. Do, J.W. Kim, S.S. Kim, J. Am. Ceram. Soc. 94, 2792–2795 (2011)
G.L. Yuan, S.W. Or, H.L.W. Chan, J. Appl. Phys 101, 064101 (2007)
M.K. Singh, S. Ryu, H.M. Jang, Phys. Rev. B 72, 132101–132104 (2005)
M. Arora, S.l Chauhan, P.C. Sati, M. Kumar, S. Chhoker, R. K. Kotnala, J. Mater. Sci. Mater. Electron. 25, 10: 4286–4299, (2014)
Acknowledgements
Authors are thankful to Department of Pure and Applied Physics, Guru Ghasidas Vishwavidyalaya for facilities generated under SAP-UGC, FIST-DST program. The author (HST) is thankful to University Grant Commission, New Delhi, India, for financial assistance through MRP (F. No. 41–954/2012(SR)). One of the authors (MD) is thankful to Guru Ghasidas Vishwavidyalaya for financial assistant through UGC scholarship. Authors are also thankful to Prof. G. D. Varma, Department of Physics, Indian Institute of Technology, Roorkee, India.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
De, M., Patel, S.P. & Tewari, H.S. Strain induced structural phase transition in NaNbO3 doped BiFeO3 . J Mater Sci: Mater Electron 28, 6928–6935 (2017). https://doi.org/10.1007/s10854-017-6393-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-6393-9