
Abstract
Eu3+ and/or Tb3+ doped NaY(WO4)2 nanomaterials have been successfully synthesized by one-step hydrothermal method. The samples were characterized by X-ray diffraction, field-emission scanning electron microscopy, X-ray energy dispersive spectroscopy, and photoluminescence spectra. The results show that the novel nanoplates with a diameter of 300–600 nm and the thickness of 20–25 nm are observed. Under the excitation of 246 or 230 nm, individual RE3+ ions activated NaY(WO4)2 phosphors exhibit excellent emission properties in their respective regions. The as-prepared Eu3+ or Tb3+ doped samples show strong red and green emission, originating from the allowed 5D0 → 7FJ (J = 0, 1, 2) transition of the Eu3+ ions and the 5D4 → 7FJ (J = 6, 5, 4, 3) transition of the Tb3+ ions. In addition, by properly tuning the relative concentration of Eu3+ ions in the case of Eu3+ and Tb3+ co-doped systems, tunable emissions in a single component are obtained under the excitation of 230 or 395 nm. Moreover, an energy transfer from Tb3+ to Eu3+ is observed, which has been justified through the luminescence spectra and the fluorescence decay curves. Furthermore, the corresponding luminescence and energy transfer mechanism have been proposed in optical transitions and possible energy transfer scheme. These results indicate that Eu3+ and Tb3+ doped NaY(WO4)2 phosphors will find potential application in the field of solid-state lighting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, inorganic luminescent materials containing rare-earth (RE) ions have become desirable in the development of optical materials. These materials have interesting optical characteristics and high potentiality in the fields of light-emitting diodes (LEDs), solid state lasers, display systems and other optoelectronic devices due to their luminescent properties largely based on the abundant emission colors deriving from the rare-earth ions [1–4]. Among them, white light-emitting diodes (W-LEDs) have been intensively studied as an attractive alternative to conventional lamps because of their excellent luminescent characteristics, good stability, high luminescence efficiency, as well as low cost [5, 6]. Phosphors are efficient luminescence materials and irreplaceable components for the development of LEDs. Recently, oxide phosphors for LEDs have gained interest due to their better thermal and chemical stability and environmental friendliness compared with sulfides, such as Y2O2S:Eu3+, Ca1−xSrxS:Eu2+ and ZnCdS:Ag red phosphors, ZnS:Cu, Al, SrGa2S4:Eu2+ green phosphors and ZnS:Ag blue phosphors [7–10], which are currently used for multicolor lighting or white lighting.
Obviously, the characteristic emissions of luminescent materials result from interactions between host and rare-earth ions used as luminescent centers. To design and achieve excellent luminescent performance, the proper host material is often indispensable. Tungstates are well-known classical self-activated luminescence materials, which can effectively absorb ultraviolet (UV) and X-ray, then simultaneously migrate the absorbed energy to the doped rare-earth ions through energy transfer, and give their characteristic emissions. Therefore, they are recently considered as promising host materials for rare-earth luminescent centers because of the special properties of WO4 2− group [11–14]. From the report of Huang et al. [15], it was seen that tungstate host essentially showed a little weaker blue emission at room temperature compared to the absorption in excitation spectrum, owing to efficient energy transfer from WO4 2− group to activators. Liu et al. [16] had also discovered that NaY(WO4)2 exhibited a broad emission centered at approximately 469 nm without doping Ln3+ ions, and the intensity of this emission decreased when Ln3+ ions were doped.
Among them, alkali rare earth tungstates (ARE(WO4)2, where A = alkali metal ions, RE = rare earth ions) are of particular interest in the potential applications in quantum electronics, visual light display system, solid-state lighting, and optoelectronic devices [17–20]. They especially had been studied widely as luminescent host in the field of solid state lasers in the past years. Furthermore, as the most frequently used and studied rare earth ions in luminescent materials, Eu3+ has been investigated primarily in red phosphors. Also, Tb3+ ions are used often for their characteristic green emission. Generally, white light emissions or individual color emissions can be achieved by systematically altering relative rare-earth ion concentration and largely depend on the energy transfer of excitation energy between rare-earth ions. Reasonable adjusting the relative dopant contents of sensitizer and activator in the case of Eu3+ and Tb3+ co-doped systems can be an effective approach to obtain multiple colors, even in some special host materials, and nearly white light for W-LEDs excited by UV light can be obtained [17–21]. Furthermore, in Eu3+ and Tb3+ co-doped systems, there is an energy transfer from Tb3+ to Eu3+ obviously, such as the unique luminescent properties of Eu3+, Tb3+ co-doped SrY2O4 [21] and Sr3AlO4F [22], the dipole–dipole type energy transfer between Tb3+ and Eu3+ in NaLa(WO4)2 [23], the energy transfer from Tb3+ to Eu3+ via its 5d and 4f states in Ba3La(PO4)3 [24] and Tb2(WO4)3:Eu3+ nanowires [25].
In order to search for a new and economical as well as high efficient phosphor, many researches have been focused on alkali rare earth tungstates. For example, Zheng et al. [12] obtained white phosphors by single doping Eu3+ in the NaY(WO4)2. Li et al. [26] prepared high brightness MLa(WO4)2:Eu3+ (M = Li, Na, K) and NaRE(WO4)2:Eu3+ (RE = Gd, Y, Lu) red phosphors. To date, tungstates doped with different rare earth ions have been prepared most maturely through high temperature solid state reaction [16, 20, 26, 27], and industrial productions of rare earth tungstates are all obtained by this method. However, there are some insurmountable defects in high temperature solid state reaction such as high temperature, long time, high energy consumption making more high-cost and depletion of energy. In consideration of this problem, the hydrothermal method is preferred because the synthesis conditions such as temperature and reaction time can be easily adjusted. Furthermore, the hydrothermal process has proved to be a facile and fast route with low cost and energy consumption, which has been widely employed for the synthesis of inorganic materials including alkali rare earth tungstates [11, 12, 28–30]. Among the alkali rare earth tungstates, the NaY(WO4)2 lattice belongs to the scheelite CaWO4 structures, which consists of two formula units in the unit cell, with space group C 64h (I41/a). Na+ and Y3+ ions are equivalent, and occupy sites of symmetry S4. Due to the entire disorder among the occupation of these atoms in their sites, for simplicity we consider them as identical. The two equivalent W6+ ions occupy sites of symmetry S4, the eight oxygen atoms are equivalent, and occupy a site of symmetry C1 [31].
In 2004, Neeraj et al. [20] prepared the NaY(WO4)2−x(MoO4)x:Eu3+ red phosphors by high temperature solid state method, which might find application in white lighting devices utilizing GaN-based excitation in the n-UV. Liu et al. [27] prepared NaY(WO4)2:Tb3+ powders by solid state reaction at 900 °C for 6 h. Also, a few more reports had been found on the synthesis of Eu3+ or Tb3+ activated NaY(WO4)2 host lattice [27–29, 32]. In our previous reports, we prepared NaLa(WO4)2:Er3+, Eu3+ phosphors [33], Tb3+ or/and Sm3+ doped NaGd(WO4)2 phosphors [34] and Tm3+, Dy3+, Eu3+ co-doped NaGd(WO4)2 phosphors [35]. However, to the best of our knowledge, no reports have been found on the Eu3+ and Tb3+ co-doped NaY(WO4)2 phosphors and the energy transfer mechanism between them in this host.
In this work, we reported the synthesis of Eu3+ and Tb3+ doped NaY(WO4)2 phosphors by a facile hydrothermal process without further sintering treatment. Moreover, we investigated the energy transfer from WO4 2− group to Eu3+ and Tb3+, respectively. In addition, by adjusting the doping concentration of Eu3+ ions in the case of Eu3+ and Tb3+ co-doped systems, an energy transfer from Tb3+ to Eu3+ was observed and tunable emission colors were realized in these materials by changing the doping concentration of Eu3+. Finally, the main mechanism for energy transfer between Tb3+ and Eu3+ in the NaY(WO4)2 phosphors was analyzed.
2 Experimental sections
2.1 Materials
Aqueous solutions of Y(NO3)3, Eu(NO3)3 and Tb(NO3)3 were obtained by dissolving the rare earth oxides Ln2O3 (Ln = Y, Eu) (99.99 %) and Tb4O7 (99.99 %) in dilute HNO3 solution (15 mol/L) under heating with agitation in ambient atmosphere. All the other chemicals were of analytic grade and used as received without further purification.
2.2 Methods
A series of rare earth-doped NaY(WO4)2 phosphors were synthesized by a facile hydrothermal process without further sintering treatment. 1.0 mmol of RE(NO3)3 [including Y(NO3)3, Eu(NO3)3 or Tb(NO3)3] were added into 10 mL of aqueous solution containing 2.0 mmol of glycine. After vigorous stirring for 20 min, 2.0 mmol of Na2WO4·2H2O was slowly added dropwise into the above solution. After additional agitation for 30 min, the resultant milky colloidal suspension was transferred to a Teflon bottle held in a stainless steel autoclave, and then heated at 260 °C for 2 h. Finally, as the autoclave was naturally cooled to room-temperature, the precipitates were separated by centrifugation, washed with deionized water and ethanol in sequence each several times, and then dried in air at 60 °C for 12 h. Specific material formula is shown in Table 1.
2.3 Characterization
The purity and phase structure of the products were examined by X-ray powder diffraction (XRD) performed on a Rigaku D/max-RA X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm) and Ni filter, operating at 20 mA, 30 kV, scanning speed, step length and diffraction range were 10°/min, 0.1° and 10°–80°, respectively. The morphology and composition of the samples were observed by a FEI XL-30 field emission scanning electron microscope (FESEM) equipped with an energy-dispersive X-ray spectrometer (EDS). The excitation and emission spectra, and the luminescence decay curves of samples were recorded with a HITACHI F-7000 Fluorescence Spectrophotometer using a Xe lamp as the excitation source, scanning at 1200 nm/min.
3 Results and discussion
3.1 Crystallization behavior and structure
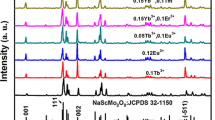
The XRD patterns of NaY(WO4)2:Eu3+, Tb3+ phosphors have been shown in Fig. 1. It is obvious that all the diffraction peaks of these samples are in good agreement with pure tetragonal NaY(WO4)2 in I41/a space group (lattice constants a = b = 5.205 Å, c = 11.251 Å, JCPDS card 48-0886) and no other phases or impurities can be detected. This result indicates that the complete crystallinity and pure-phase double alkaline rare-earth tungstates with the general formula of NaY(WO4)2 can be obtained by one-step hydrothermal method and the Eu3+ ions and Tb3+ ions are completely incorporated into the NaY(WO4)2 host lattice without making significant changes to the crystal structure. Moreover, the high crystallinity is still obtained by low temperature. Typically, the doped ions occupy sites in the host largely depending on the effective radii (r) of the cations. The effective radii of the Tb3+ ions (0.1044 nm for CN = 8) and the Eu3+ ions (0.1066 nm for CN = 8) are similar to those of the Y3+ ions (0.1019 nm for CN = 8). In view of the effective radii, Tb3+ and Eu3+ ions are expected to occupy the Y3+ sites preferentially because the radii of the Na+ ions (0.118 nm) are too large for Tb3+ and Eu3+ ions as well as the valence state analysis.

X-ray powder diffraction patterns of NaY (WO4)2:Eu3+, Tb3+ phosphors
3.2 Morphology and composition
The morphology and composition features of NaY(WO4)2:0.03Eu3+ phosphor were investigated by FESEM and EDS, as shown in Figs. 2a–c. The FESEM images reveal that it is composed of regular nanoplates with the diameter from 300 to 600 nm and the thickness ranging from 20 to 25 nm (Fig. 2a, b). The plate-like morphology formation may be resulted from the addition of glycine, which is acted as a surfactant to control the shape of the samples, the similar role can be seen in other Ref. [36]. EDS pattern shown in Fig. 2c performs the chemical composition of the product, containing Na, Y, Eu, W, and O (silicon and gold signals are from silicon host and spraying gold process). Combined with the above XRD patterns, the samples are further proved to be NaY(WO4)2.

FESEM images (a, b) and EDS pattern (c) of NaY (WO4)2:0.03Eu3+ phosphor
3.3 Photoluminescence properties
Room temperature photoluminescence spectra of the synthesized NaY(WO4)2 nanocrystals doped with Ln3+ were investigated. Figure 3 shows the PL and PLE spectra of NaY(WO4)2:Tb3+, NaY(WO4)2:Eu3+, and NaY(WO4)2:Eu3+, Tb3+ phosphors. It is well known that the excitation spectrum of NaY(WO4)2:0.02Tb3+ (monitored at 545 nm) consists of two components. The former is a strong and broad band from 200 to 300 nm with a maximum at about 230 nm due to f-d transitions of Tb3+ and O2−–W6+ charge transfer transitions within the WO4 2− groups, which was deconvolved into two bands using Gaussian fitting shown in Fig. 4a. The peak located at 226 nm is due to the f-d transitions of Tb3+ and the peak centered at 264 nm is due to the CTB of WO4 2−. The latter is the weak f-f transitions of Tb3+ in the longer wavelength region, whose are hardly detected. They are located at 341, 352, 359, 369, 378 and 488 nm corresponding to the transitions from the 7F6 ground state to the different excited states of Tb3+, namely, 5G2, 5D2, 5G5, 5G6, 5D3 and 5D4, respectively, which are shown in Fig. 3c. The PL spectrum of the 2 mol% Tb3+ activated NaY(WO4)2 nanocrystals by exciting at 230 nm is shown in Fig. 3a (right). It can be seen that there are two groups of emissions. The first broad band from 410–540 nm with a maximum at about 450 nm belongs to the blue emission of WO4 2− group. The second group of emission bands locates at 488 nm in the blue region and 545 nm in the green region, which belong to 5D4 → 7F6 and 5D4 → 7F5 transitions of Tb3+, respectively. Compared Fig. 3a, b, it can be seen that for NaY(WO4)2:Tb3+ phosphors, with a lower doping concentration of Tb3+ (shown in Fig. 3a), the blue emission of WO4 2− centered at 450 nm is strong. As Tb3+ concentration increases, the emission of WO4 2− is depressed owing to the more energy transfer from WO4 2− group to Tb3+, which enhances the transitions of 5D4 to 7Fj of Tb3+ with a green emission (Fig. 3b, right). Figure 3d shows the PLE and PL spectra of NaY(WO4)2:0.03Eu3+ phosphor. The PLE spectrum monitored with 616 nm emission (5D0–7F2) of Eu3+ reveals the broad band from 200 to 310 nm, which is charge transfer band (CTB) caused by the charge transfer from the completely filled 2p6 orbital of O2− ions to the partially filled 4f6 orbital of the Eu3+ ions as well as the charge transfer between O2− and W6+, which indicates the energy transfer from WO4 2− groups to Eu3+ ions [27, 37, 38]. For a clear understanding, the broad band was also deconvolved with Gaussian fitting, which has been shown in Fig. 4b. The peak located at 234 nm is due to the CTB of Eu3+–O2− and the peak centered at 262 nm is due to the CTB of WO4 2−. There are also some sharp excitation peaks at the long wavelength in the PLE spectrum of Eu3+ doped NaY(WO4)2. They correspond to f-f transitions of Eu3+ assigned to the transitions of (7F0–5D4) at 363 nm, (7F0–5L7) at 385 nm, (7F0–5L6) at 395 nm, (7F0–5D3) at 416 nm, (7F0–5D2) at 466 nm. These excitation peaks indicate that the phosphor can strongly absorb UV, violet and blue light to obtain Eu3+ emission. Upon excitation with 246 nm, NaY(WO4)2:Eu3+ phosphor exhibits a board blue emission attributed to the transition of WO4 2− in short wavelength region and some high intense emission peaks corresponding to the transitions of Eu3+ in long wavelength region, whose positions are at 579 nm (5D0–7F0), 591 nm (5D0–7F1), 616 nm (5D0–7F2), respectively. The strongest emission is observed due to the 5D0–7F2 electric dipole transition (at 616 nm), which is about 5.64 times stronger than that of the magnetic dipole transition 5D0 → 7F1 (at 591 nm), indicating that Eu3+ ions are positioned at the sites without inversion symmetry in the NaY(WO4)2 host [30, 39]. Furthermore, in Fig. 3d, it can be clearly seen that there is a significant spectral overlap between the broad WO4 2− emission band and several excitation bands of Eu3+ ions located at 416 and 466 nm, indicating a favourable condition for possible energy transfer from WO4 2− group to Eu3+ ions. This interesting phenomenon can also be observed in NaY(WO4)2:Tb3+ sample in both Fig. 3a, b, confirming the energy transfer between WO4 2− group and Tb3+ ions. However, comparing the energy transfer between WO4 2− and Eu3+ to that between WO4 2− and Tb3+, it is well known that the latter is much more efficient (Fig. 3b, d). This standpoint has also been reported by Liao et al. [40]. Figure 3e illustrates the PLE and PL spectra of NaY(WO4)2:0.03Eu3+, 0.02Tb3+ phosphor. It can be noted that in the PLE spectrum, with the different monitoring wavelengths (545 nm for Tb3+ and 616 nm for Eu3+), the broad band from 200 to 300 nm monitored by 545 nm is much stronger than that of monitored by 616 nm, further confirming that the more efficient energy transfer between WO4 2− and Tb3+ (Fig. 3e, left). In the PL spectra of NaY(WO4)2:0.03Eu3+, 0.02Tb3+, it is observed both red characteristic emission and green characteristic emission and the emission intensity of Eu3+ is considerably enhanced compared to that of single Eu3+ doped sample (Fig. 3d), while the emission intensity of Tb3+ is distinctly decreased (Fig. 3b) because of energy transfer from Tb3+ to Eu3+. The energy transfer between Tb3+ and Eu3+ can be confirmed by excitation spectrum of NaY(WO4)2:0.03Eu3+, 0.02Tb3+ sample monitored at 616 nm (Fig. 5). There is an excitation peak at about 488 nm (transition 7F6 → 5D4 of Tb3+). In emission spectrum of Tb3+ excited at 488 nm, there is no peak in range of 600–625 nm in NaY(WO4)2:0.02Tb3+ (solid line in Fig. 5b), but in NaY(WO4)2:0.03Eu3+, 0.02Tb3+ (dash line in Fig. 5b) there is a peak of Eu3+ at 616 nm. Thus, emission intensity of Eu3+ excited at 488 nm is attributed to energy transfer from Tb3+ to Eu3+.

PL and PLE spectra of NaY(WO4)2:0.02Tb3+ (a); NaY(WO4)2:0.03Tb3+ (b); expansion in 325–500 nm for PLE spectra of NaY(WO4)2:0.03Tb3+ (c); NaY(WO4)2:0.03Eu3+ (d); NaY(WO4)2:0.03Eu3+, 0.02Tb3+ (e) phosphors

Gaussian fitting curves of the broad band from 200 to 300 nm for the NaY(WO4)2:0.03Tb3+ (a) and NaY(WO4)2:0.03Eu3+ (b) phosphors

PLE spectra of NaY(WO4)2:0.03Eu3+, 0.02Tb3+ (λem = 545 and 616 nm) and NaY(WO4)2:0.03Eu3+ (λem = 616 nm) (a); PL spectra of NaY(WO4)2:0.03Eu3+, 0.02Tb3+ (dash line) and NaY(WO4)2:0.02Tb3+ (solid line) (λex = 488 nm) (b)
In order to understand the energy transfer process, a series of samples were prepared. The Tb3+ mole content was fixed at 0.02 and the doping molar concentration of Eu3+ was varied from 0.001 to 0.03. Figure 6 depicts the variation of PL spectra and emission intensity of Tb3+ and Eu3+ in the NaY(WO4)2:xEu3+, 0.02Tb3+ samples with varying molar concentration of Eu3+ under excitation at 230 nm. It is noted that the emission intensities of Tb3+ (5D4 → 7F6 at 488 nm, 5D4 → 7F5 at 545 nm) decrease remarkably while the PL intensities of Eu3+ (5D0 → 7F2 at 616 nm) gradually increase with increasing Eu3+ molar concentration from 0.001 to 0.03, indicating the enhancement of energy transfer from the Tb3+ ions to Eu3+ ions.

Series of PL spectra of NaY(WO4)2:xEu3+, 0.02Tb3+ (x = 0, 0.001, 0.005, 0.0075, 0.01, 0.02, 0.03) under UV excitation (λex = 230 nm) (a); dependence of the emission intensity at different wavelength on Eu3+ concentration (b)
To further validate the process of energy transfer, the fluorescence lifetimes τ for Tb3+ (5D4 → 7F5 at 545 nm) with different Eu3+ concentrations are measured and presented in Fig. 7. The decay behaviour of Tb3+ can be expressed as
with the Eu3+ concentration changing from 0.005 to 0.03 in the NaY(WO4)2:xEu3+, 0.02Tb3+ phosphors, the integrated intensities of Tb3+ emissions also monotonically decrease from 0.9015 times to 0.5144 times as that of NaY(WO4)2:0.02Tb3+. The line shown the relationship between the fluorescence lifetimes of the Tb3+ and Eu3+ doping concentration tends to be a non-exponential function with increasing Eu3+ concentration (Fig. 7, Inset), strongly demonstrating the characteristics of energy transfer between Tb3+ and Eu3+. As reported, the luminescence intensities of various rare earth ions can be enhanced or quenched by the energy transfer from other co-doped rare earth ions [22]. Moreover, it is well-known that an effective energy transfer between Tb3+ and Eu3+ can take place in several hosts, such as molybdates [41], yttria [42], and tungstates [43]. The energy-transfer efficiency ηT can be calculated using the following formula [44]:
where τ and τ0 are the lifetimes for Tb3+ with and without the Eu3+ ions. The energy-transfer efficiency is calculated as a function of Eu3+ concentrations and is shown in the inset of Fig. 7. It can be seen that the efficiency η T increases with increasing Eu3+ concentration and reaches 7.44861 % at x = 0.03. This is because the probability of the energy transfer from Tb3+ to Eu3+ is proportional to R−6 (R is the average distance between Tb3+ and Eu3+). With the increase of Eu3+ concentration, the average distance (R) between Tb3+ and Eu3+ is reduced, so the energy transfer efficiency is increased [22].

Decay curves for the luminescence of Tb3+ ions in NaY(WO4)2:xEu3+,0.02Tb3+ samples. (excited at 230 nm, monitored at 545 nm). Inset dependence of the fluorescence lifetime of the Tb3+ and energy transfer efficiency ηT on Eu3+ doping concentration in NaY(WO4)2:xEu3+, 0.02Tb3+ phosphors
A schematic model proposed for the probable ways of energy transfer in Tb3+ and Eu3+ co-doped NaY(WO4)2 is shown in Fig. 8. During the excitation process, the electrons situated at oxygen 2p states absorb energies of photons from UV. As consequence of this phenomenon, the energetic electrons are promoted to tungsten 5d states located near the conductor band [45, 46]. When the electrons fall back to lower energy states again via blue emission and energy transfer to Eu3+ and Tb3+ ions, and some energy is lost by cross relaxation [47]. Furthermore, the 5D4–7F6, 5D4–7F5 or 5D4–7F4 (5D4–7F3) emissions of Tb3+ overlap well with the absorption bands 7F0–5D2 (7F0–5D1), 7F0–5D1 or 7F0–5D0 of Eu3+, respectively, and thus the energy transfer also occurs between Tb3+ and Eu3+ through the cross-relaxation process. In order to obtain the multicolor light emission, it is necessary to control the energy transfer efficiency by selecting a suitable excitation wavelength for Tb3+ and Eu3+ co-doped samples. So we chose the board band area as excitation wavelength.

Schematic energy-level diagram showing the excitation and emission mechanism of NaY(WO4)2:Eu3+, Tb3+ phosphors (ET energy transfer; NR non radiative)
Generally, the energy transfer from a sensitizer to an activator in a phosphor system may take place via exchange interaction or multipolar interaction. Based on Dexter’s energy transfer formula of multipolar interaction and Reisfeld’s approximation, the following equation can be used to analyze the potential mechanism:
in which η so and η s is the intrinsic luminescence quantum efficiency of the Tb3+ ions without and with the existence of activator Eu3+; and C is the doped concentration of Eu3+. The values of ηso/ηs can be approximately calculated by the ratio of related luminescence intensities (Iso/Is), where I so is the intrinsic luminescence intensity of Tb3+, and I s is the luminescence intensity of Tb3+ in the presence of Eu3+. When the value of n is 6, 8 or 10, the interaction corresponds to dipole–dipole, dipole–quadrupole, or quadrupole–quadrupole, respectively. The Iso/Is plots are illustrated in Fig. 9 and the plots are used linear fitting. It can be clearly seen that when n = 6, linear fitting result is the best, implying that energy transfer from Tb3+ to Eu3+ occurs via a dipole–dipole mechanism.

Dependence of Iso/Is of Tb3+ on a C 6/3Eu × 104, b C 8/3Eu × 105, and c C 10/3Eu × 106 in the NaY(WO4)2:xEu3+, 0.02Tb3+ phosphors
We have studied the CIE values of the suite of NaY(WO4)2:xEu3+, 0.02Tb3+ phosphors. The CIE chromaticity coordinates for the phosphors excited at 230 and 395 nm have been calculated, which are represented in the CIE diagram of Fig. 10. For the NaY(WO4)2:Eu3+, Tb3+ phosphors, when excited at 230 nm, the trend of their color tones changes from blue to white by adjusting the doping concentration of Eu3+, the corresponding chromaticity coordinates are (0.1799, 0.2716), (0.1818, 0.2590), (0.1842, 0.2558), (0.2078, 0.2910) and (0.2244, 0.2805), respectively. Furthermore, the color tone of the phosphors shifts gradually from white (which is represented at point 7) to red with exciting at 395 nm. The corresponding chromaticity coordinates are listed in Table 2. Therefore, from Fig. 10 and Table 2, it can be well seen that multiple colors are achieved by reasonable adjusting the doping concentration of Eu3+ in co-doped phosphors or through altering properly different excitation wavelengths. Moreover, the white-light emission is obtained using 395 nm as pumping source, which can find potential for WLEDs.

CIE chromaticity diagram of the selected NaY(WO4)2:Eu3+, Tb3+ phosphors under 230 nm (point 1–5), 246 nm (point 6) and 395 nm (point 7–11) excitations
4 Conclusions
In summary, Eu3+ and/or Tb3+ doped NaY(WO4)2 nanoplates were prepared by one-step hydrothermal method at 260 °C for 2 h. The as-prepared Eu3+ or Tb3+ doped samples show strong red and green emission. And the energy transfer of WO4 2− to Tb3+ is more efficient. In addition, in the case of Eu3+ and Tb3+ co-doped systems, an efficient and strong energy transfer occurs from Tb3+ to Eu3+ via a dipole–dipole mechanism. And the photoluminescence emission color tones change from greenish blue to pink by different excitation wavelengths, which may make them excellent candidates in solid-state lighting. Particularly, white light emission is exhibited with 395 nm irradiation potentially finding applications in WLEDs.
References
W.B. Im, Y.I. Kim, H.S. Yoo, D.Y. Jeon, Luminescent and structural properties of (Sr1−x, Bax)3MgSi2O8:Eu2+: effects of Ba content on the Eu2+ site preference for thermal stability. Inorg. Chem. 48, 557–564 (2009)
Z. Wang, G. Li, Z. Quan, D. Kong, X. Liu, M. Yu, J. Lin, Nanostructured CaWO4, CaWO4:Pb2+ and CaWO4:Tb3+ particles: polyol-mediated synthesis and luminescent properties. J. Nanosci. Nanotechno. 7, 602–609 (2007)
G. Blasse, B.C. Grabmaier, Luminescent Materials (Springer, Berlin, 1994)
L.L. Beecroft, C.K. Ober, Nanocomposite materials for optical applications. Chem. Mater. 9, 1302–1317 (1997)
T. Hashimoto, F. Wu, J.S. Speck, S. Nakamura, A GaN bulk crystal with improved structural quality grown by the ammonothermal method. Nat. Mater. 6, 568–571 (2007)
R. Mueller-Mach, G. Mueller, M.R. Krames, H.A. Höppe, F. Stadler, W. Schnick, T. Juestel, P. Schmidt, Highly efficient all-nitride phosphor-converted white light emitting diode. Phys. Status Solidi A 202, 1727–1732 (2005)
M. Kottaisamy, K. Horikawa, H. Kominami, T. Aoki, N. Azuma, T. Nakamura, Y. Nakanishi, Y. Hatanaka, Synthesis and characterization of fine particle Y2O2S:Eu red phosphor at low-voltage excitation. J. Electrochem. Soc. 147, 1612–1616 (2000)
K. Murakami, H. Kudo, T. Taguchi, M. Yoshimo, Compound semiconductor lighting based on InGaN ultraviolet LED and ZnS phosphor system, in 2000 IEEE International Symposium on Compound Semiconductors (2000), pp. 449–454
Y. Hu, W. Zhuang, H. Ye, S. Zhang, Y. Fang, X. Huang, Preparation and luminescent properties of (Ca1−x, Srx)S:Eu2+ red-emitting phosphor for white LED. J. Lumin. 111, 139–145 (2005)
Y. Sato, N. Takahashi, S. Sato, Full-color fluorescent display devices using a near-UV light-emitting diode. Jpn. J. Appl. Phys., Part 2 35, L838–L839 (1996)
F. Lei, B. Yan, H.H. Chen, J.T. Zhao, Surfactant-assisted hydrothermal synthesis of Eu3+-doped white light hydroxyl sodium yttrium tungstate microspheres and their conversion to NaY(WO4)2. Inorg. Chem. 48, 7576–7584 (2009)
Y. Zheng, H. You, K. Liu, Y. Song, G. Jia, Y. Huang, M. Yang, L. Zhang, G. Ning, Facile selective synthesis and luminescence behavior of hierarchical NaY(WO4)2:Eu3+ and Y6WO12:Eu3+. CrystEngComm 13, 3001–3007 (2011)
J. Liu, J.M. Cano-Torres, C. Cascales, F. Esteban-Betegón, M.D. Serrano, V. Volkov, C. Zaldo, M. Rico, U. Griebner, V. Petrov, Growth and continuous-wave laser operation of disordered crystals of Yb3+:NaLa(WO4)2 and Yb3+:NaLa(MoO4)2. Phys. Status Solidi A 202, R29–R31 (2005)
A. Garcia-Cortes, C. Cascales, A. de Andres, C. Zaldo, E.V. Zharikov, K.A. Subbotin, S. Bjurshagen, V. Pasiskevicius, M. Rico, Raman scattering and Nd3+ laser operationin NaLa(WO4)2. IEEE J. Quantum Electron. 43, 157–167 (2007)
S. Huang, X. Zhang, L. Wang, L. Bai, J. Xu, C. Li, P. Yang, Controllable synthesis and tunable luminescence properties of Y2(WO4)3:Ln3+ (Ln = Eu, Yb/Er, Yb/Tm and Yb/Ho) 3D hierarchical architectures. Dalton Trans. 41, 5634–5642 (2012)
X. Liu, W. Xiang, F. Chen, Z. Hu, W. Zhang, Synthesis and photoluminescence characteristics of Dy3+ doped NaY(WO4)2 phosphor. Mater. Res. Bull. 48, 281–285 (2013)
Y.S. Zhao, H. Fu, F. Hu, A.D. Peng, J. Yao, Multicolor emission from ordered assemblies of organic 1D nanomaterials. Adv. Mater. 19, 3554–3558 (2007)
A.A. Kaminskii, H.J. Eichler, K.-I. Ueda, N.V. Klassen, B.S. Redkin, L.E. Li, J. Findeisen, D. Jaque, J. García-Sole, J. Fernández, R. Balda, Properties of Nd3+-doped and undoped tetragonal PbWO4, NaY(WO4)2, CaWO4, and undoped monoclinic ZnWO4 and CdWO4 as laser-active and stimulated raman scattering-active crystals. Appl. Opt. 38, 4533–4547 (1999)
Y.R. Do, Y.D. Huh, Optical properties of potassium Europium tungstate phosphors. J. Electrochem. Soc. 147, 4385–4388 (2000)
S. Neeraj, N. Kijima, A.K. Cheetham, Novel red phosphors for solid-state lighting: the system NaM(WO4)2−x (MoO4) x :Eu3+ (M = Gd, Y, Bi). Chem. Phys. Lett. 387, 2–6 (2004)
E. Pavitra, G.S.R. Raju, Y.H. Ko, J.S. Yu, A novel strategy for controllable emissions from Eu3+ or Sm3+ ions co-doped SrY2O4:Tb3+ phosphors. Phys. Chem. Chem. Phys. 14, 11296–11307 (2012)
M. Shang, G. Li, X. Kang, D. Yang, D. Geng, J. Lin, Tunable Luminescence and Energy Transfer properties of Sr3AlO4F:RE3+ (RE = Tm/Tb, Eu, Ce) Phosphors. ACS Appl. Mater. Interfaces 3, 2738–2746 (2011)
Y. Tian, B. Chen, B. Tian, N. Yu, J. Sun, X. Li, J. Zhang, L. Cheng, H. Zhong, Q. Meng, R. Hua, Hydrothermal synthesis and tunable luminescence of persimmon-like sodium lanthanum tungstate:Tb3+, Eu3+ hierarchical microarchitectures. J. Colloid Interface Sci. 393, 44–52 (2013)
C. Zhang, H. Liang, S. Zhang, C. Liu, D. Hou, L. Zhou, G. Zhang, J. Shi, Efficient sensitization of Eu3+ emission by Tb3+ in Ba3La(PO4)3 under VUV-UV excitation: energy transfer and tunable emission. J. Phys. Chem. C 116, 15932–15937 (2012)
Z. Hou, Z. Cheng, G. Li, W. Wang, C. Peng, C. Li, P.A. Ma, D. Yang, X. Kang, J. Lin, Electrospinning-derived Tb2(WO4)3:Eu3+ nanowires: energy transfer and tunable luminescence properties. Nanoscale 3, 1568–1574 (2011)
L. Li, L. Liu, W. Zi, H. Yu, S. Gan, G. Ji, H. Zou, X. Xu, Synthesis and luminescent properties of high brightness MLa(WO4)2:Eu3+ (M = Li, Na, K) and NaRE(WO4)2:Eu3+ (RE = Gd, Y, Lu) red phosphors. J. Lumin. 143, 14–20 (2013)
X. Liu, W. Xiang, F. Chen, W. Zhang, Z. Hu, Synthesis and photoluminescence of Tb3+ activated NaY(WO4)2 phosphors. Mater. Res. Bull. 47, 3417–3421 (2012)
S. Huang, D. Wang, Y. Wang, L. Wang, X. Zhang, P. Yang, Self-assembled three-dimensional NaY(WO4)2:Ln3+ architectures: hydrothermal synthesis, growth mechanism and luminescence properties. J. Alloys Compd. 529, 140–147 (2012)
N. Xue, X. Fan, Z. Wang, M. Wang, Mater. Lett. 61, 1576 (2007)
F. Wang, X. Fan, D. Pi, Z. Wang, M. Wang, Hydrothermal synthesis and luminescence behavior of rare-earth-doped NaLa(WO4)2 powders. J. Solid State Chem. 178, 825–830 (2005)
S. Perets, R.Z. Shneck, R. Gajic, A. Golubovic, Z. Burshtein, Vibrational spectra of sodium gadolinium tungstate NaGd(WO4)2 single crystals: observation of spatial dispersion. Vib. Spectrosc. 49, 110–117 (2009)
X. Qian, X. Pu, D. Zhang, L. Li, M. Li, S. Wu, Combustion synthesis and luminescence properties of NaY1−xEux(WO4)2 phosphors. J. Lumin. 131, 1692–1695 (2011)
Y. Liu, Y.X. Liu, G.X. Liu, X.T. Dong, J.X. Wang, Up/Down conversion, tunable photoluminescence and energy transfer properties of NaLa(WO4)2:Er3+, Eu3+ phosphors. RSC Adv. 5, 97995–98003 (2015)
Y. Liu, Y.X. Liu, G.X. Liu, X.T. Dong, J.X. Wang, W.S. Yu, Luminescence, energy transfer and tunable color properties of a single-component Tb3+ or/and Sm3+ doped NaGd(WO4)2 phosphors with UV excitation for WLEDs. RSC Adv. 4, 58708–58716 (2014)
Y. Liu, Y.X. Liu, G.X. Liu, J.X. Wang, X.T. Dong, W.S. Yu, A single component and warm-white-emitting phosphor NaGd(WO4)2:Tm3+, Dy3+, Eu3+: synthesis, luminescence, energy transfer and tunable color. Inorg. Chem. 53, 11457–11466 (2014)
Y.Y. Jiang, Y. Liu, G.X. Liu, X.T. Dong, J.X. Wang, W.S. Yu, Q. Dong, Surfactant-assisted hydrothermal synthesis of octahedral structured NaGd(MoO4)2:Eu3+/Tb3+ and tunable photoluminescent properties. Opt. Mater. 36, 1865–1870 (2014)
P. Jia, X. Liu, Y. Luo, M. Yu, J. Lin, Sol–Gel synthesis and characterization of SiO2@NaGd (WO4)2:Eu3+ core-shell-structured spherical phosphor. J. Electrochem. Soc. 154, J39–J43 (2007)
C.H. Chiu, M.F. Wang, C.S. Lee, T.M. Chen, Structural, spectroscopic and photoluminescence studies of LiEu(WO4)2−x(MoO4)x as a near-UV convertible phosphor. J. Solid State Chem. 180, 619–627 (2007)
N. Xue, X. Fan, Z. Wang, M. Wang, Synthesis process and the luminescence properties of rare earth doped NaLa(WO4)2 nanoparticles. J. Phys. Chem. Solids 69, 1891–1896 (2008)
J. Liao, S. Zhang, H. You, H.-R. Wen, J.L. Chen, W. You, Energy transfer and luminescence properties of Eu3+-doped NaTb(WO4)2 phosphor prepared by a facile hydrothermal method. Opt. Mater. 33, 953–957 (2011)
A. Khanna, P.S. Dutta, Narrow spectral emission CaMoO4:Eu3+, Dy3+, Tb3+ phosphor crystals for white light emitting diodes. J. Solid State Chem. 198, 93–100 (2013)
D. Tu, Y. Liang, R. Liu, D. Li, Eu/Tb ions co-doped white light luminescence Y2O3 phosphors. J. Lumin. 131, 2569–2573 (2011)
M.V. Nazarov, D.Y. Jeon, J.H. Kang, E.J. Popovici, L.E. Muresan, M.V. Zamoryanskaya, B.S. Tsukerblat, Luminescence properties of europium–terbium double activated calcium tungstate phosphor. Solid State Commun. 131, 307–311 (2004)
P.I. Paulose, G. Jose, V. Thomas, N.V. Unnikrishnan, M.K.R. Warrier, Sensitized fluorescence of Ce3+/Mn2+ system in phosphate glass. J. Phys. Chem. Solids 64, 841–846 (2003)
J.C. Sczancoski, L.S. Cavalcante, N.L. Marana, R.O. da Silva, R.L. Tranquilin, M.R. Joya, P.S. Pizani, J.A. Varela, J.R. Sambrano, M.S. Li, E. Longo, J. Andrés, Electronic structure and optical properties of BaMoO4 powders. Curr. Appl. Phys. 10, 614–624 (2010)
V.M. Longo, L.S. Cavalcante, R. Erlo, V.R. Mastelaro, A.T. de Figueiredo, J.R. Sambrano, S. de Lázaro, A.Z. Freitas, L. Gomes, N.D. Vieira Jr., J.A. Varela, E. Longo, Strong violet-blue light photoluminescence emission at room temperature in SrZrO3: joint experimental and theoretical study. Acta Mater. 56, 2191–2202 (2008)
A.M. Li, D.K. Xu, H. Lin, S.H. Yang, Y.Z. Shao, Y.L. Zhang, Z.Q. Chen, Facile morphology-controllable hydrothermal synthesis and color tunable luminescence properties of NaGd(MoO4)2:Eu3+, Tb3+ microcrystals. RSC Adv. 5, 45693–45702 (2015)
Acknowledgments
This work was supported by the National Natural Science Foundation of P.R. China (NSFC) (Grant Nos. 51072026, 51573023) and the Development of Science and Technology Plan Projects of Jilin Province (Grant No. 20130206002GX).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, Y., Liu, G., Wang, J. et al. Hydrothermal synthesis, multicolor tunable luminescence and energy transfer of Eu3+ or/and Tb3+ activated NaY(WO4)2 nanophosphors. J Mater Sci: Mater Electron 27, 10780–10790 (2016). https://doi.org/10.1007/s10854-016-5183-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-016-5183-0