
Abstract
Nanocrystalline alumina–zirconia powders were prepared by a modified chemical route using sucrose, polyvinyl alcohol (PVA) and metal nitrates followed by a post calcination process. The process involved dehydration of Al3+–Zr4+ ions-sucrose–PVA solution to a highly viscous liquid which on decomposition process produced a black precursor material. The obtained precursor were then calcined at various temperatures: 1,050, 1,100, 1,150, 1,200 and 1,250 °C for different soaking times (1, 2, 4 h) in air. The formation of a nanocomposite composed of α-alumina (~20 nm) and tetragonal (t) zirconia (~19 nm) crystallites were confirmed for the sample calcined at 1,200 °C for 2 h, based on our XRD and TEM results. However, for the samples calcined below 1,150 °C the composite formed were composed of metastable alumina (γ, δ, θ) as well as t-zirconia phases. Interestingly, the zirconia phase retained its tetragonal structure for all the samples calcined above 1,050 °C. This is possibly related to the “size effect” and reduction of surface enthalpy of the zirconia crystallites surrounded by Al3+ cations.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Synthesis of nano-sized advanced engineering ceramics and other specialty materials is one of the major challenges in material processing technology [1, 2]. Nanocrystalline materials show superior phase homogeneity, enhanced sinterability at a relatively low temperature and microstructures leading to unique electrical, mechanical, dielectric, magnetic and optical properties [3]. Tetragonal zirconia in an alumina matrix is known as zirconia toughened alumina (ZTA). ZTA enjoys both the advantage of combination of low cost alumina and high strength of zirconia. High density sintered bodies of ZTA have shown interesting mechanical properties [4]: fracture toughness of 7 MPa m1/2, hardness of 15 GPa and flexural strength of 910 MPa [5] making them as a novel candidate for the applications in the areas of extreme mechanical abrasion and thermal shock such as machining steels and cast irons [6]. The fracture toughness and the strength of alumina ceramics are improved remarkably by the dispersion of 10–20 vol% of meta-stable tetragonal (t) zirconia particles which can transform into the stable monoclinic phase (m) under loading. In such a case, if a crack grows through the ceramic, the zirconia particles in the region of the crack change from (t) to monoclinic (m) phase with a martensitic transformation which is accompanied by a volume expansion of about 3%. This will generate compressive stresses in the alumina matrix thus contrasting the opening of the advancing crack giving rise to increased toughness. Therefore, one can realize the important role of the presence of t-zirconia on service life of these composites. However, other mechanisms such as crack deflection, crack bridging and the presence of micro cracks may also enhance the toughness [7, 8].
Various synthesis routes such as conventional dry/wet milling [9, 10] of the mixture of zirconia and alumina, sol–gel [11], liquid precursor method [12] have been already reported for the synthesis of ZTA. It is worth to mention that oxide complex solid solutions prepared by conventional solid state synthesis route (also referred as mixed oxide route) normally suffer from the lack of homogeneity at atomic scale, poor sinterability and possible introduction of extrinsic impurities during milling process. These mentioned drawbacks can be mostly overcome by utilizing chemical synthesis methods such as classic sol–gel method using alkoxide precursors. Though, such routes also in most cases introduce problems of moisture sensitivity, high costs of the initial raw materials and the low production yield of the process. Pati et al. [13] developed a novel and inexpensive chemical route for the synthesis of nano-sized α-Al2O3 powder using sucrose and polyvinyl alcohol (PVA) and aluminum metal-nitrate. This preparation route involves dehydration of the solution of metal ion with sucrose and PVA followed by decomposition of the polymer matrix. A voluminous organic based fluffy black mass was thus obtained after complete dehydration of the resulting solution. This dried carbonaceous product was then ground to obtain fine powders forming the precursor material. Upon appropriate calcination of this precursor nano-sized alumina powder was obtained [13]. We in this work, however, for the first time report the synthesis and characterization of alumina–zirconia nanocomposite powder (0.15 Al2O3–0.85 ZrO2 in molar ratios) by this method [13].
2 Experimental
2.1 Chemicals and preparation of the initial raw materials
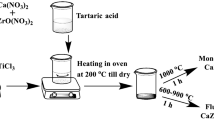
The initial chemicals used were Al(NO3)3 · 9 H2O (>99.8% purity Merck), zirconium oxychloride octa hydrate (99.99% purity, Aldrich Chemicals), nitric acid (Aldrich Chemicals) polyvinyl alcohol (PVA, MW 125000, Aldrich Chemicals) ammonium hydroxide (Aldrich Chemicals) and sucrose (MW 342, Merck). For preparing zirconium precursor solution initially zirconium oxychloride was dissolved in a minimum quantity of distilled water. Zirconium hydroxide was then precipitated by the addition of ammonium hydroxide to this solution. This was subsequently washed with distilled water several times to remove alkalines. The zirconium hydroxide product thus obtained was then reacted by appropriate amounts of nitric acid to get zirconium nitrate solution.
2.2 Synthesis of ZTA
For the synthesis of ZTA precursor, the appropriate amount of Al(NO3)3 · 9 H2O was dissolved in distilled water and then added to the already prepared zirconium nitrate solution. Further, the solutions of 10% (w/v) PVA and 40% (w/v) sucrose in distilled water were also separately prepared. Then appropriate amounts of the metal nitrates solution was then mixed with sucrose solution such that the metal ions to sucrose mole ratio could be maintained at 1:4. Moreover, in this solution, 10% PVA (w/v) was also subsequently mixed maintaining the ratio of metals ions to vinyl alcohol monomer unit at 1:0.7. The pH of the final solution was kept in between 3 and 4 by adding few drops of HNO3. A highly acidic sucrose solution can self polymerize or cross-link with PVA to form a 3-dimensional network [14]. The final thus obtained clear solution was then evaporated on a hot plate at 180 °C while stirring. This gave rise to the formation of a black fluffy product which could act as a fuel during calcination process. The black precursor product later ground to obtain a fine powder. In order to decarbonize and remove the organic materials from the precursor, all of the sample were kept at 500 °C for 4 h before subjecting them to the final calcination step at: 900, 950, 1,000, 1,050, 1,100, 1,150, 1,200 and 1,250 °C for various soaking times (1/4, 1, 2 h) in ambient air atmosphere. Various techniques such as simultaneous thermal analysis (STA), NETZSCH STA 409 PC, X-ray diffraction (XRD), PHILIPS EXPERT-PRO, Fourier transform infra-red (FTIR) analysis, SHIMADZU IR Solution, 8400S and transmission electron microscopy (TEM), PHILIPS (CM 200) 200 kV were utilized to characterize the samples prepared. The crystallite sizes of the different phases were determined from the full-width at half maximum (FWMH) of their corresponding strongest diffraction peaks using the Williamson–Hall method [15] after applying the standard correction for instrumental broadening.
3 Results and discussion
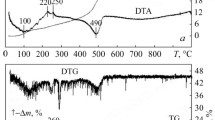
Figure 1 displays the STA result obtained for the prepared black precursor material at a heating rate of 10 °C/min. As is evident from this figure the observed mass loss between room temperature to ~200 °C is due to the desorption of the surface adsorbed H2O molecules and/or interstitial gases during the processing.

STA analysis of the prepared black precursor at a heating rate of 10 K/min
Evolution of various gases such as CO, CO2, NH3 and NO2 are common during the early stage of heat-treatment process for a polymer precursor with metal cations in acidic medium. The appearance of the endothermic peak below 200 °C also supports the desorption process. The rate of weight loss was, however, considerably increased between ~315 and 600 °C temperature range for which a broad exothermic peak of two maxima at ~370 and 575 °C could be detected (Fig. 1). The increased rate of weight loss and the appearance of the broad exothermic peak are related to the decomposition and combustion of the precursor powder. The appearance of the broad exo-peak could be attributed to the removal of different organic materials available in the synthesized ZTA precursor of different chemical natures. No further weight loss was observed for the temperatures above 603 °C. Furthermore, the two small exothermic peaks appeared at ~875 °C and ~1,150 °C are possibly related to alumina phase transformations as will be discussed later (Fig. 1).
Figure 2 illustrates the FTIR multiplot pattern of the non-calcined black precursor as well as those calcined at 500 and 1,250 °C for 2 h.

FTIR analysis of the non-calcined black precursor (a) as well as those calcined at 500 °C (b) and 1,250 °C (c) for 2 h
The appearance of the absorption peaks between ~420 and 700 cm−1 for the sample calcined at 1,250 °C are possibly related to the Al–O and Zr–O bonds formed. The broad peak centered at ~3,200 cm−1 detected for the initial precursor and the sample calcined at 500 °C is attributed to the hydroxyl group which noticeably disappeared for the sample calcined at 1,250 °C. The FTIR spectrum of the black precursor powder revealed the strong absorption bands at around 1,400 cm−1 and 1,600 cm−1 which are possibly attributed to the various vibrational modes of the carboxyl group [16] formed due to the oxidation of sucrose and PVA with nitrate ions. Further, the appearance of the bands between 800 and 1,050 cm−1 for the non-calcined precursor is possibly related to the presence of some non decomposed nitrate ions. Increasing the calcination temperature gave rise to a noticeable decrease of the intensities or disappearance of most of the absorption peaks related to various organic bonds formed in the range ~800–3,000 cm−1 (Fig. 2). Figure 3 demonstrates the XRD multiplot of the non calcined sample as well as those calcined at various temperatures at a fixed soaking time of 2 h. The presence of mainly an amorphous structure could be confirmed for both initial black precursor and the sample calcined at 500 °C (Fig. 3).

XRD multiplot patterns of various samples calcined at different temperatures for a soaking time of 2 h as well as non calcined one. All of these samples were decarbonized at 500 °C for 4 h before being subjected to the final calcination step
However, increasing the calcination temperature to 900 °C gave rise to the early signs of the formation of various alumina metastable phases (Fig. 3) in agreement with our STA results. XRD pattern of the sample calcined at 1,000 °C revealed the formation of t-zirconia phase along with other alumina meta-stable phases such as: γ, δ, θ. Partial conversion of meta-stable alumina phase/s to stable α-alumina took place for the sample calcined at 1,150 °C (Fig. 3), as confirmed by our XRD results. The observed phase conversion fully accomplished for the samples calcined at 1,200 and 1,250 °C for whose the formation of α-Al2O3 and t-zirconia phases could be verified (Fig. 3).
Boehmite (AlO(OH)) is the most commonly used precursor for preparing α-Al2O3. It is already shown that during heat treatment the aluminum oxide hydroxide undergoes the following series of polymorphic phase transformations before final conversion to the thermodynamically stable phase i.e. α-Al2O3 polymorph (corundum): boehmite → γ → δ → θ → α [17]. The θ- to α-Al2O3 phase transformation is considered to be performed by a nucleation and growth mechanism [18]. The free energy of α-Al2O3 is always lower than those of transitional phases. The nucleation and the crystallite growth processes of α-Al2O3 in a θ-matrix are mainly composed of three successive and distinct processes: primary recrystallization, grain growth and secondary crystallization [19]. During heat treatment process, the θ-Al2O3 crystallite grows exceeding the critical size needed for the formation of a stable α-Al2O3 nucleus [20, 21] and the nucleation of α-Al2O3 nuclei occurs. Continuous heating lead to the growth of α-Al2O3 crystallites and the phase formation is accomplished. For the samples prepared in this work, we assume that a similar phase transformation is possibly taking place for the alumina phase. However, this demands further study which is currently under way. Furthermore, it is interesting to note that for all of the samples calcined between 1,000 and 1,250 °C the crystalline structure of the observed zirconia phase was tetragonal. ZrO2 has three polymorphs of P2 1 /c monoclinic (m), P4 2 /nmc tetragonal (t), and Fm3m cubic (c) crystal structures, with m-zirconia the equilibrium bulk structure at room temperature [22]. Bulk m-zirconia transforms to t- or c-ZrO2 at 1,170 or 2,370 °C, respectively. Various attempts have been made to stabilize the c- or t-ZrO2 to exist at room temperature by doping small amounts of different dopants such as Y2O3, MgO, CaO, CeO2, AlN, Mg3N2, Si3N4, Al2O3 [23–27]. These additives are believed to control the c- or t-ZrO2 nucleation and growth in small crystallites through their total surface strain/free energy [25, 28, 29]. It should be also added that the process depends on the solubility of these dopants in c- or t-zirconia and the chemistry of the stabilizer used. Some of the mentioned dopants segregate to form grain boundaries and part of them substitute in Zr4+ lattice sites. The latter gives rise to lattice strain due to cation size mismatch and creation of oxygen vacancies to compensate the electronic charges and hence increasing the total energy of the system required for the room temperature stabilization of the high energy meta-stable c- or t-zirconia phases. In general, three forms of stabilized zirconia are generally seen: a fully stabilized one containing only c-ZrO2 phase, a partially stabilized ZrO2 containing t-zirconia grains in c-ZrO2 matrix, and the third one the so-called t-ZrO2 polycrystals comprising of fine t-ZrO2 grains [30]. It is already shown that in Al3+-doped zirconia the stabilization of the zirconia can be both related to “size effect” [31] and the presence of aluminum ions on the surface of the zirconia particles [32]. The latter is believed [33] to reduce the surface enthalpy and hence increases the stability region of the tetragonal phase. In order to determine the effect of the presence of alumina matrix on the phase stability of zirconia phase, we also prepared pure zirconia powder with similar preparation route applied for the rest of the samples described in the experimental section. Figure 4 displays XRD pattern of the pure zirconia powder decarbonized at 500 °C for 4 h followed by calcination at 1,250 °C for 2 h. As is clear from this figure, the formation of a fully monoclinic phase with average crystallite size of 40 nm was obtained for this sample. This confirms the crucial effect of the presence of the matrix alumina phase on the structure and crystallite size of the zirconia phase.

XRD pattern of the pure zirconia precursor sample decarbonized at 500 °C for 4 h followed by calcination at 1,250 °C for 2 h
Figure 5 a–c demonstrate the TEM bright field images of the sample calcined at 1,150 °C for 2 h at different magnifications as well as their corresponding selected area diffraction pattern (SADP).

a–c TEM bright field images of the sample calcined at 1,150 °C for 2 h at different magnifications as well as their corresponding SADP. The dark appearing crystallites in this picture present zirconia phase and the lighter one belong to alumina phase
The dark crystallites in these micrographs present the zirconia phase and the lighter one represents alumina phase. As can be noticed, the zirconia crystallites are rather uniformly distributed throughout the matrix. Further, the presence of an interconnecting amorphous interface phase was also confirmed for this sample evidenced by the appearance of its corresponding hallo diffraction pattern (shown as the inset in Fig. 5c). The rather complex diffraction pattern obtained for the sample calcined at 1,150 °C for 2 h (Fig. 5b) was found to be related to the presence of various alumina transitional phases (γ, δ, θ) along with the rhombohedral α-Al2O3 phase in agreement with our XRD results (Fig. 3).
The crystallite sizes of α-alumina and t-zirconia phases in the case of the sample calcined at 1,200 °C for 2 h were ~20 and 19 nm, respectively. A two-fold increase in the average size of α-alumina crystallites was detected for the sample calcined at 1,250 °C (~36 nm) compared to that of the sample calcined at 1,150 °C (~18 nm). However, a slower growth rate was comparatively detected for the t-ZrO2 crystallites. The average sizes of t-ZrO2 crystallites were ~17 and 23 nm for the samples calcined at 1,150 and 1,250 °C, respectively. The existence of a slower crystallite growth in case of t-ZrO2 could be related to the reduction of the surface enthalpy of this phase which can give rise to a slower sintering kinetics and grain growth rate during the calcination process. Furthermore, the average crystallite sizes of the t-zirconia phase for the samples calcined at 950, 1,000 and 1,050 °C for a fixed soaking times of 2 h were ~8.5, 9 and 9.5 nm, respectively. For these samples, such a slow crystallite size growth is possibly related to the presence of amorphous alumina phase surrounding the zirconia phase acting as a diffusion barrier during the calcination process at these rather low temperatures. Aluminum ions segregated at high temperatures in the grain boundaries of zirconia highly doped with alumina [34] is also reported to show a similar behavior.
For the samples decarbonized at 500 °C for 4 h and calcined at 1,250 °C increasing the final calcination soaking time from 1/4 to 2 h gave rise to the increase of crystallite sizes of α-alumina and t-zirconia phases. The crystallite sizes of α-alumina phase for the samples calcined at 1,250 °C for 1/4, 1 and 2 h were ~28, 31 and 36 nm, respectively. The magnitudes of the crystallite sizes of t-zirconia phase were ~20, 22 and 24 nm for the samples kept for 1/4, 1 and 2 h at 1,250 °C, respectively. The rise of soaking time from 1/4 to 4 h also caused the increase of the corresponding diffraction peak intensities of these two phases suggesting higher level of formation of α-alumina and t-zirconia phases in these samples (Fig. 6). However, for the sample calcined at 1,250 °C for 1/4 h the existence of small amounts of metastable alumina phases could be detected as shown in Fig. 6.

XRD patterns of the samples calcined at 1,250 °C for various soaking periods
4 Conclusions
In summary successful synthesis of α-Al2O3 and t-zirconia nanocomposite is reported here by a modified sol–gel method using sucrose and PVA. The nanocomposite powder was prepared by thermal decomposition of a sucrose–PVA solution containing Al3+ and Zr4+ cations followed by calcination at various temperatures and soaking periods. Samples calcined at 1,200 °C for 2 h revealed the formation of α-alumina (~20 nm) and t-zirconia (~19 nm) phases. However, for the samples calcined below 1,150 °C the presence of metastable alumina phases (γ, δ, θ) besides t-zirconia could be confirmed. Further, rather sharp rise of α-Al2O3 crystallite sizes was seen for the samples calcined at 1,250 °C (~36 nm) compared to that of the samples calcined at 1,150 °C (~18 nm). However, this was not the case for the t-zirconia phase: ~17 nm (1,150 °C, 2 h), ~23 nm (1,250 °C, 2 h).
References
G.H. Beall, L.R. Pinckney, J. Am. Ceram. Soc. 82, 5 (1999)
H. Gleiter, Acta Mater. 48, 1 (2000). doi:10.1016/S1359-6454(99)00285-2
H. Singh Nalwa, Nanostructured materials and nanotechnology (Academic Press, London, 2002)
T. Sornakumar, M.V. Gopalakrishnan, R. Krishnamurphy, C.V. Gokularathnam, Int. J. Refract. Metab. Hard Mater. 13, 375 (1995). doi:10.1016/0263-4368(95)00026-F
Y. Murase, E. Kato, K. Daimon, J. Am. Ceram. Soc. 69, 195 (1986). doi:10.1111/j.1151-2916.1986.tb04706.x
B. Mondal, A.B. Chattopadhyay, A. Virkar, A. Paul, Wear 156, 365 (1992). doi:10.1016/0043-1648(92)90229-2
W.H. Tuan, R.Z. Chen, T.C. Wang, C.H. Cheng, P.S. Kuo, J. Eur. Ceram. Soc. 22, 2827 (2002). doi:10.1016/S0955-2219(02)00043-2
F.L. Matthews, R.D. Rawlings, Composite materials: engineering and science (Chapman and Hall, London, 1994), p. 137
B. Kibbel, A.H. Heuer, J. Am. Ceram. Soc. 69, 231 (1986). doi:10.1111/j.1151-2916.1986.tb07414.x
D.J. Green, J. Am. Ceram. Soc. 65, 610 (1982). doi:10.1111/j.1151-2916.1982.tb09939.x
B. Fegley Jr., P. White, H.K. Bowen, J. Am. Ceram. Soc. 68, C-60 (1985)
M.L. Balmer, F.F. Lange, V. Jayram, C.G. Levi, J. Am. Ceram. Soc. 78, 1489 (1995)
R.K. Pati, J.C. Ray, P. Pramanik, Mater Lett. 44, 299 (2000)
R.N. Das, Mat. Lett. 47, 344 (2001)
G.K. Williamson, W.H. Hall, Acta Metall. 1, 22 (1953)
J.A. Gadsden, Infrared spectra of minerals and related inorganic compounds (Butterworth, London, 1975), p. 193
K. Wefers, C. Misra, Oxides and hydroxides of aluminium, (ALCOA Technical pap No. 19 Rev., ALCOA Labs, 1987)
F.W. Dynys, J.W. Halloran, J. Am. Ceram. Soc. 65, 442 (1982)
W.D. Kingery, H.K. Bowen, D.R. Uhlmann, Introduction to ceramics, 2nd edn. (Wiley, New York, 1976)
B. Djuricic, S. Pickering, P. Glaude, D. Mcgarry, P. Tam-Buyser, J. Mater. Sci. 32, 589 (1997)
J.R. Wynnyckyj, C.G. Morris, Metall. Trans. B 16, 345 (1985)
R.D. Purohit, S. Saha, A.K. Tyagi, Mater. Sci. Eng. B 130, 57 (2006)
S. Moreau, M. Geravais, A. Douy, Solid State Ionics 625, 101–103 (1997)
N. Claussen, R. Wanger, L.J. Gauckler, G. Petzor, J. Am. Ceram. Soc. 61, 369 (1978)
Y. Murase, E. Kato, K. Diamon, J. Am. Ceram. Soc. 69, 83 (1986)
K. Ishida, K. Hirota, O. Yamaguchi, H. Kume, S. Inamura, H. Miyamoto, J. Am. Ceram. Soc. 77, 1319 (1994)
S. Kikkawa, A. Kijima, K. Hirota, O. Yamamoto, J. Am. Ceram. Soc. 85, 721 (2002)
R.C. Garvie, J. Phys. Chem. 69, 1238 (1965)
A.H. Heuer, N. Claussen, W.M. Kriven, M. Ruhle, J. Am. Ceram. Soc. 65, 642 (1982)
A.H. Heuer, J. Am. Ceram. Soc. 70, 689 (1987)
R.C. Garvie, J. Am. Chem. Soc. 82, 218 (1978)
S. Ram, A. Mondal, Appl. Surf. Sci. 221, 237 (2004)
V.V. Srdic, M. Winterer, A. Moller, G. Miehe, H. Hahn, J. Am. Ceram. Soc. 84(12), 2771 (2001)
V.V. Srdic, M. Winterer, H. Hahn, J. Am. Ceram. Soc. 83, 1853 (2000)
Acknowledgments
We would like to acknowledge the help of Nanomaterials Group, Advanced Materials Research Center (AMRC), IUST. Further, the support of Iranian Nanotechnology Initiative Council (INIC) is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Beitollahi, A., Hosseini-Bay, H. & Sarpoolaki, H. Synthesis and characterization of Al2O3–ZrO2 nanocomposite powder by sucrose process. J Mater Sci: Mater Electron 21, 130–136 (2010). https://doi.org/10.1007/s10854-009-9880-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-009-9880-9