
Abstract
The influences of oxygen vacancies on the electronic properties of V2O5−x have been investigated by photoelectron spectroscopy and first principle calculations. Photoelectron spectroscopic data suggested that the vanadium (V) ions are gradually reduced to lower oxidation states with the increases of oxygen (O) vacancy density. Simultaneously, the formation of O vacancies leads to a decrease in the work function of V2O5−x . Theoretical calculations further prove that the formation of O vacancies would cause a reduction in the charge state of V ions and a decrease of work function. The electronic structures of V ions are strongly modified by the removal of O ions nearby due to electron transfer to the 3d orbitals of the V ions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since it is a promising material in many technological applications, vanadium pentoxide (V2O5) has been the subject of intense work due to its small and highly anisotropic n-type electrical conductivity [1–7]. Especially, α-V2O5 is a widely used industrial catalyst for a variety of chemical reactions such as the oxidation of hydrocarbons and sulfur dioxide [8, 9]. The oxidation involves Mars-van Krevelen [10] redox cycles using lattice oxygen (O) as reactive intermediates [11]. The ability of vanadium (V) atoms to possess multiple stable oxidation states results in the easy conversion between vanadium oxides of different stoichiometry by oxidation or reduction and is believed to be an important factor leading to variations in its electronic properties. Alteration of electronic properties of V2O5 will strongly affect its usefulness. It is generally considered that O vacancies will form as point defect in the V2O5 lattice when V2O5 is simply annealed in a vacuum chamber or exposed to reducing environments at elevated temperatures. Removal of a neutral O atom may result in trapping of an electron in the formed cavity thus changing the electronic structure of the resulting V2O5−x . The effects of the formation of surface O vacancies on the electronic structures of V2O5−x have been previously studied theoretically by Ganduglia-Pirovano et al. [12, 13]. In present work, the effect of bulk O vacancies on the electronic properties of α-V2O5−x was investigated by a combination of X-ray and ultraviolet photoelectron spectroscopy (XPS and UPS) studies and density functional theory (DFT) calculations.
2 Experimental details and calculation method
A V2O5 thin film (sample a) was first deposited on a freshly cleaved highly orientated pyrolytic graphite substrate at room temperature (RT) from a homemade physical vapor deposition effusion cell using a BN crucible in an ultrahigh vacuum (UHV) chamber. Reduced V2O5−x thin films were subsequently formed by annealing the as-prepared V2O5 thin film in the UHV chamber at 200 and 300 °C for 1 h (samples b and c, respectively). The XPS and UPS experiments were carried out at RT with a surface analysis system (Phi 5700). The base pressure during the measurements was better than 1 × 10−9 mbar. Al Kα radiation (hν = 1486.6 eV) from a monochromatized X-ray source was used for XPS. A UV light (hν = 21.22 eV) from a discharge lamp was employed for UPS measurements. The spectra are given as the binding energy (BE) referred to the Fermi level of a sputter cleaned Ag reference sample. Sample stoichiometry ratios S i,j are calculated from the XP spectra using the following formula [1]:
where C i and C j are the concentrations of the elements, I i and I j the background corrected intensities of the photoelectron emission lines and ASF i and ASF j the atomic sensitivity factors for photoionization of the ith and jth elements. For example the O to V ratio (S O,V ) in stoichiometric V2O5 should be 2.5.
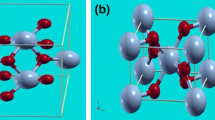
DFT was employed for electronic structure calculations utilizing the Vienna Ab-initio Simulation package (VASP) in a local density approximation (LDA). VASP is a complex package for performing ab-initio quantum-mechanical molecular dynamic simulations with plane-wave basis sets and ultra-soft Vanderbilt pseudopotentials. The electron-core interaction is described by the projector augmented wave (PAW) method [12]. For the defect calculation we use a supercell with 28 atoms equivalent to 1 × 1 × 2 primitive unit cell. Based on the literature [12, 14], we removed one or two vanadyl oxygen (O1) ions from V8O20 thus forming V8O19 or V8O18 as shown in Fig. 1a. In this calculations a non-shifted Γ-point centered 4 × 4 × 4 k-point mesh was employed. The geometry of the V2O5 lattice has been optimized by varying the c value whilst remaining the ratios of c/a and c/b. The lattice constants of a = 11.497 Å, b = 3.559 Å and c = 4.335 Å, which are only about 0.7% relative error compared with those lattice constants reported previously, have been obtained.

The crystal structures of (a) V8O20, (b) V8O19 and (c) V8O18; the location of V1, V2, V3, V4 and O1 ions are illustrated
3 Results and discussion
Figure 2 shows the XP spectra of the O 1s and V 2p core level emission lines for samples a, b and c. The BEs of O 1s and V 2p 3/2 are 529.6 and 516.8 eV for sample a, which are consistent with the literature values for stoichiometric V2O5 [15]. But a very small shoulder is found at the lower BE side of V 2p 3/2 indicating a small amount of vanadium in the reduced +4 oxidation state. Spectra b and c show that the V 2p 3/2 and V 2p 1/2 lines become broader and shift to lower BEs. The decrease in the BE of the core level (chemical shift) usually indicates a decrease in the positive charge of the transition metal atoms. Simultaneously, the O 1s lines of spectra b and c become asymmetric. The intensity ratios (S O,V ) of O 1s / V2p 3/2 calculated according to Eq. 1 are listed in Table 1. The S O,V for sample a is about 2.48, which suggests that 4% of V ions are in the +4 oxidation state, i.e., the O vacancy concentration is about 4%. The S O,V for samples b and c are 2.35 and 2.10. The decrease of the S O,V implies the decomposition of V2O5 after heating at higher temperatures in vacuum. The removal of the originally negatively charged O ions as neutral species leads to the reduction of the V ions. Originally empty 3d orbitals of V ions adjacent to a vacancy are able to localise the excess electrons left by the removed O ions, leading to the formation of V4+ in the vicinity of the vacancy. Further details based on the UPS data and the theoretical calculations are discussed later. Figure 3 reports the UP spectra measured from the same samples as those shown in Fig. 2. In these spectra, there are three distinct lines between 3 and 8 eV, they exhibit a well-defined valence band shape with binding energies at about 3.7, 5.4 and 6.9 eV, respectively. It is clear that a broad V 3d signal appears at ∼1.3 eV and increases in the order of a to c. This implies that the V 3d orbital is gradually occupied due to the increase of the O vacancy concentration, as more and more V ions are reduced to lower oxidation species. Further, from the UP spectra, the change of work functions has been noted. The work function could be calculated as hν (21.22 eV)−BESEO (where BESEO is the BE of secondary emission onset indicated in Fig. 3). It decreases with increasing O vacancy concentration. This phenomenon is possibly due to the concentration change of reduced V species on the surface. The higher the concentration of V4+ species leads to a decrease in the work function.

XP spectra of O 1s and V 2p core levels of V2O5−x for samples a, b and c

HeI (21.2 eV) spectra of samples a, b and c
In the theoretical calculation, we found that the total energy of V8O20, V8O19 and V8O18 are about −253.8, −243.9 and −234.2eV, respectively, which means that the formation of O vacancies will lower the stability. The total density of states (DOSs) of (a) V8O20, (b) V8O19 and (c) V8O18 are showed in Fig. 4. Compared with V8O20, the valence band minima of V8O19 and V8O18 shift to lower energies. This result is consistent with the decrease of work function found on the UP spectra for chemical nonstoichiometric V2O5−x . In the conduction bands, it is obvious that the split-off in V8O20 caused by the crystal field into lower (t2g) and upper (eg) segments [16] disappears in V8O19 and V8O18. The conduction band consists mainly of V 3d state [17]. This indicates that the V 3d orbital is strongly influenced by the formation of the O vacancy. Shown in Fig. 5 are the conduction band structures of (a) V8O20, (b) V8O19 and (c) V8O18. The energy bands that are degenerated in V8O20 split off in V8O19 and V8O18. The conduction band structure of V8O19 shows an additional energy band at the bottommost indicated by the arrow. It is so close to the conduction band that we could not find it in the total DOSs. It exactly matches the donor level caused by O vacancy [14]. Consequently, the V8O18 shows two donor levels at the bottom of the conduction band. These results suggest again that the electrons left by the O vacancy would locate in V 3d orbitals. In order to understand the effect of an O vacancy on the oxidation state of V ions, the partial charges (q/e) for different V ions are summarized in Table 2; the locations of different V ions are labeled in Fig. 1. The original charge of V ions in V8O20 is about +2.64 q/e, indicating that there is strong electronic hybridization between V and O ions [17]. When one O vacancy is formed, the average charge of V ions decreases to +2.58 q/e, showing that an O vacancy would donate about 0.48 q/e (8×(2.64−2.58)) to the V ions. It is not surprising that the V1 ion closest to an O vacancy is reduced seriously from 2.64 to 2.14. The partial charges of V2, V3 and V4 ions hardly change, indicating that the removal of O only affects the electron charge of the nearest V ion and has very little influence on the V ions far away from it. The average charge of V ions in V8O18 is about 0.14 q/e less than that in V8O20. Therefore, 1.1 electrons are transferred to V ions caused by two O vacancies. In Fig. 1c, both of the partial charges of V1 and V2 ions decrease, while the partial charges of the V3 and V4 ions almost does not change. It is generally considered that the O vacancy is the basic point defect in the V2O5 lattice. Its formation may be described by the following equations expressed in Kröger-Vink notation:

The DOSs of (a) V8O20, (b) V8O19 and (c) V8O18

The conduction band structures of (a) V8O20, (b) V8O19 and (c) V8O18
where V O denotes the O vacancy, ° and ′ are the positive and negative charges with respect to the lattice. Removal of an O atom may result in trapping of an electron in the formed cavity [18]. The electron left behind after the removal of a neutral O atom from the perfect crystal can be localized at the ions around the cavity. However, there is a finite possibility of them being trapped and localized in the center of the vacancy. This has been proved by the above experimental and theoretical studies: the oxidation state of the nearest-neighbor V ions to the O vacancy is reduced and its electronic structures are altered seriously. One O vacancy will only transfer 0.48 electrons to the closest V ions, the rest would still be localized in the center of the vacancy [18].
4 Conclusions
The influences of the O vacancies on the electronic structures of V2O5−x have been investigated by a combination of photoemission spectroscopy study and theoretical calculations. The O vacancy defect electrons will become partially localised at the V ions nearby and cause them to experience a decrease in their partial charge; one O vacancy would transfer about 0.48 electrons to nearest V 3d states. Moreover, the formation of O vacancies would decrease the work function of V2O5−x due to the formation of lower oxidation state V ions.
References
Q.-H. Wu, A. Thissen, W. Jaegermann, Surf. Sci. 578, 203 (2005)
Q.-H. Wu, A. Thissen, W. Jaegermann, Solid State Ionics 167, 155 (2004)
Q.-H. Wu, A. Thissen, W. Jaegermann, M. Liu, Appl. Surf. Sci. 236, 473 (2004)
Q.-H. Wu, A. Thissen, W. Jaegermann, Appl. Surf. Sci. 250, 57 (2005)
Q.-H. Wu, A. Thissen, W. Jaegermann, Appl. Surf. Sci. 252, 1801 (2005)
Q.-H. Wu, A. Thissen, W. Jaegermann, Appl. Surf. Sci. 253, 1713 (2006)
Q.-H. Wu, Surf. Interface Anal. 38, 1179 (2006)
M.A. Banares, Catal. Today 51, 319 (1999)
B.M. Weckhuysen, D.E. Keller, Catal. Today 78, 25 (2003)
P. Mars, D.W. van Krevelen, Spec. Suppl. Chem. Eng. Sci. 3, 41 (1954)
K. Chen, E. Iglesia, A.T. Bell, J. Catal. 192, 197 (2000)
M.V. Ganduglia-Pirovano, J. Sauer, Phys. Rev. B 70, 045422 (2004)
M.V. Ganduglia-Pirovano, J. Sauer, J. Phys. Chem. B 109, 374 (2005)
S. Laubach, P.C. Schmidt, A. Thißen, F.J. Fernandez-Madrigal, Q.-H. Wu, W. Jaegermann, M. Klemm, S. Horn, Phys. Chem. Chem. Phys. 9, 2564 (2007)
G.A. Sawatzky, D. Post, Phys. Rev. B 20, 1546 (1979)
D.W. Bullett, J. Phys. C 13, L595 (1980)
Q.-H. Wu, A. Thissen, W. Jaegermann, M. Schuz, P.C. Schmidt, Chem. Phys. Lett. 430, 309 (2006)
A.M. Ferrati, G. Paccioni, J. Phys. Chem. 99, 17010 (1999)
Acknowledgements
We acknowledge the financial support of this work by National Natural Science Foundation of China (NO:20603028), the Project of Young Talents Innovation of Fujian Province (NO: 2005J005) and the Project of New Century Excellent Talent of Fujian Province.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, ZY., Wu, QH. The effects of oxygen vacancies on the electronic properties of V2O5−x . J Mater Sci: Mater Electron 19 (Suppl 1), 366–370 (2008). https://doi.org/10.1007/s10854-007-9506-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-007-9506-z