
Abstract
A novel hybrid porous carbon has been synthesized by a facile method blending chitosan with gelatin in acetic acid solution. Gelatin changes the morphology of the chitosan-derived carbon materials and produces abundant micro- and mesopores with the help of KHCO3. More importantly, gelatin brings heteroatoms and pseudo-capacitance into hybrids to improve the capacitive performance as the nitrogen source. A unique tangerine pith-like morphology comes into being on the chitosan-derived hybrid porous carbon, and there are lots of interconnected micropores forming with high specific surface area of 927.17 m2 g−1. This particular morphology, high nitrogen content and specific surface area ensure the as-obtained carbon electrode wonderful capacitive performance with specific capacitance of 331 F g−1 in 6 mol L−1 KOH electrolyte at 1 A g−1, high retention of 73% and excellent capacity stability of 90% after 10000 cycles at 10 A g−1. Additionally, the chitosan-derived hybrid porous carbon shows remarkable energy density of 34 W h kg−1 with the power density of 900 W kg−1, superior to majority of the commercial devices. The simple and efficient strategy and the excellent electrochemical performance are of great significance to the large-scale application of biomass-based supercapacitors.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As potential energy storage units, supercapacitors (SCs) have attracted extensive attention and been widely applied in electric vehicles, medical devices and forklifts, due to their long cycle life, quick charge and discharge, high power density, and low maintenance cost [1,2,3]. Currently, commercial products are primarily based on porous carbon using ion adsorption, for example, carbon nanotubes [4], mesoporous carbon [5], activated carbon [6], graphitic carbon [7]. Meanwhile, carbon combined with conductive polymers or transition-metal hydroxides/oxides can greatly increase energy storage capacity over a period of time through offering pseudo-capacitance by reversible redox reactions on the interface between the electroactive material and electrolyte [8,9,10]. The inherent capacitance of the carbon component is very important due to the fact that most of the pseudo-capacitive materials have the problems of inferior conductivity, poor cyclic stability, and high cost [11]. Moreover, porous carbon-based SCs electrode materials are known to face sluggish electrode kinetics relating to internal pore ion transport and transfer [12,13,14]. For instance, activated carbon has a long diffusion path (> 5 μm) and a large electrolyte ion-transfer resistance, which result in a large voltage drop (IR) and a small electrolyte ion-approachable specific surface area (SSA) at the large current density [15, 16]. The carbon nanotubes have remarkable conductivity and high electrolyte ion-approachable SSA that can offer ideal rate stability [17]; however, the value of electrical double-layer capacitance is restricted. Therefore, there it is an enormous and urgent challenge to design and develop a new carbonaceous electrode material with high performances for SCs.
The energy storage of carbon-based material is mainly based on the adsorption of ions; meanwhile, the surface charges are equally distributed at the interfaces between the electrode and the electrolyte [18]. Hence, high SSA and abundant pore structures adapted to solute/solvent ion sizes are required for carbon-based SCs electrode materials [19,20,21]. Additionally, incorporation of surface functional groups and heteroatom species (e.g., nitrogen (N), sulfur (S), phosphorus (P) and boron (B)) can modify the surface wettability of carbonaceous materials and provide pseudo-capacitive behavior [22,23,24,25]. Such doping effects can obviously increase the performances of SCs [26, 27].
In recent years, the synthesis of carbonaceous materials derived from biomass for energy applications has become a research focus due to the increasing lack of mineral fuels [28]. Biomass materials, such as foods, agricultural wastes, animal bones, silks, and even natural polymers, have been used as carbon source. For example, activated carbons fabricated through pyrolysis of popcorn have a specific capacitance (Csp) value of 245 F g−1 [29]. High-performance plant leaves-derived carbon showed a Csp value of 400 F g−1 and an energy density (Ecell) value of 55 W h kg−1 in 1 mol L−1 H2SO4 aqueous solution [30]. Raymundo et al. [31] reported the synthesis of microporous carbon materials deriving from natural seaweeds. The obtained carbon materials showed a Csp value of 264 F g−1 in 1 mol L−1 H2SO4 aqueous solution. Compared with usual carbon precursors (e.g., coal, pitch and phenolic resins), the superiorities of non-pollution and low cost facilitate the progress of biochar-based high-performance SCs in practical applications.
Chitosan, as natural amino polysaccharide, recently has been applied extensively in the field of chemical industry and food processing because of its rich resources and favorable biocompatibility. Furthermore, chitosan can be modified chemically, due to the existence of massive amino group (–NH2) and hydroxyl group (-OH) which make it a potential carbonaceous precursor with good electrochemical performances. For example, Lota et al. [32] carbonized chitosan with KOH directly and the as-prepared activated carbon materials possess 295 F g−1 at 0.1 A g−1. At the same time, Zhang et al. [33] fabricated chitosan aerogel with 3D interconnected hierarchical porous structure by crosslinking of chitosan and glutaraldehyde with Csp value 246.5 F g−1 at 0.5 A g−1. Beyond that Ling et al. [34] reported a sheet-shaped chitosan-based carbon gel and the Csp value can be as high as 242 F g−1 at the current density of 0.1 A g−1. Therefore, we expect the renewable and inexpensive chitosan can be a useful carbon precursor to achieve novel carbon-based electrode materials with outstanding capacitive performance.
In current work, the hybrid porous carbon was successfully synthesized through activating the mixture of chitosan and gelatin, which were prepared by a simple solution blending and freeze-drying process. The gelatin contains many kinds of functional groups (such as –OH, –NH2, and –COOH) on the main chains that can directly bring heteroatoms into the carbon skeleton and thereby improve the electrochemical performance of chitosan-based carbon electrode materials. Furthermore, gelatin can not only change the microstructure of chitosan-based carbon materials, but also increase the porosity and SSA of chitosan-based carbon materials as a pore-forming agent. The as-prepared hybrid porous carbon exhibits a novel sheet with tangerine pith-like nanowires evenly distributed on its surface, interconnected porous structure and high SSA of 927.17 m2 g−1. Consequently, it shows excellent energy storage capacity of 331 F g−1 at 1 A g−1, good cycle stability with 90% retention after 10000 cycles at 10 A g−1, and high energy density of 34 W h kg−1 at the power density of 900 W kg−1 in 6 mol L−1 KOH electrolyte. Compared with most chitosan-based carbon materials, the current research work has greatly enhanced the structural characteristics and energy storage performances.
Experimental section
Chemicals
Chitosan (80–95%, degree of deacetylation), gelatin and hydrochloric acid (HCl, 36–38%) used were purchased from Sinopharm Chemical Reagent Co., Ltd. Potassium bicarbonate (KHCO3, ≥ 99.5%) and acetic acid (≥ 99.5%) were supplied by Tainjin Baishi Chemical Industry Co. Ltd. All the chemicals were of analytical reagent and used without further processing.
Synthesis of CHPCs
Chitosan (2.0 g) was initially added into 100 mL 0.3 M acetic acid aqueous solution and magnetically stirred at 50 °C for 0.5 h until completely dissolved. Subsequently, gelatin (0.50 g) was dissolved into the above solution and stirred for 1 h to form a homogenous solution. Then the well-mixed solution was frozen overnight and freeze-dried into a sponge-like composite which was marked as chitosan-derived carbonaceous precursor. The precursor was calcined at 300 °C in Ar flowing for 3 h with a heating rate of 2 °C min−1 and further ground into powder after cooling naturally. The powder (0.20 g) was immersed in 5 mL KHCO3 solution (mpowder:mKOH = 1:1) and dried at 40 °C to form the mixture of powder and KHCO3. Next, the mixture was carbonized at 600 °C with the same heating conditions and washed with 0.5 M HCl solution to remove the impurities. The obtained sample was washed several times with deionized water and ethanol and collected by centrifugation and dried at 40 °C for 6 h. For comparison systematically, the control samples were prepared following the same experiment strategy with different dosages of gelatin (0, 0.25, 0.50, 0.75, and 1.0 g) or without the addition of chitosan. The final products were denoted as CPC, CHPC-0.25, CHPC-0.50, CHPC-0.75, CHPC-1.0 and GPC, respectively.
Materials characterization
The scanning electron microscope (SEM, Zeiss G500) and transmission electron microscope (TEM, JEM 2100) were used to characterize the morphological changes of the samples. The N2 adsorption–desorption measurements were measured by the specific surface aperture analyzer (Geminiv 2380) after vacuum drying for 24 h at 180 °C. The pore-size distribution and the pore parameters were analyzed by original density functional theory (ODFT) and Brunauer–Emmett–Teller (BET), respectively. The X-ray diffraction patterns (XRD) were carried out on the X-ray powder diffractometer (XRD-6100, Cu source) at the scan rate of 5 min−1. And the Raman spectra were evaluated on the LabRAM HR Evolution with solid-state laser of 532 nm and ranged from 600 to 3600 cm−1. The thermos-gravimetric analysis (TGA) was tested on the SDTA851e with the temperature range of 45–800 °C in the atmosphere N2. The X-ray photoelectron spectroscopy (XPS) was analyzed by using ESCALABXi + with the Al/Kα as the monochromatic source.
Electrochemical measurements in 6 M KOH electrolyte
The preparation processes of the working electrode are as follows: the samples, acetylene black and polyvinylidene fluoride (PVDF) were mixed into slurry in the N-methyl-2-pyrrolidone at the weight ratio of 8:1:1, then the mixture was coated on the stainless steel mesh and dried at 45 °C for 10 h (1 cm × 1 cm, about 2.5 mg). All the electrochemical tests, such as galvanostatic charge–discharge (GCD), cyclic voltammetry (CV), and electrical impedance spectroscopy (EIS), were carried out on a CHI660E electrochemical workstation in the 6 mol L−1 KOH electrolyte. The GCD and CV texts varied the constant current from 0.5 to 10 A g−1 and the scan rate from 10 to 100 mV s−1 within the voltage range of − 0.2 to 1.2 V, respectively. The frequency range for the EIS was from 0.01 to 105 Hz with the open-circuit potential.
In the typical three-electrode system (half-cell), platinum plate and a standard mercuric oxide electrode were served as the counter electrode and reference electrode, respectively. The specific capacitance (Csp, F g−1) of the electrode can easily be deduced by GCD curves based on Eq. (1) [35]:
In the symmetrical two-electrode system (full-cell), two electrodes with the same mass were back to back and separated by the polypropylene membrane. Similarly, the specific capacitance (Ccell, F g−1) was also calculated according to Eq. (2) [36]:
The energy density (Ecell, W h kg−1) and power density (Pcell, W kg−1) were obtained through Eqs. (3) and (4) [35]:
where I is the discharge current (A), Δt represents the discharge time (s), ΔV refers to the voltage window (V), and msp and mcell (g) are the mass of active materials on a single electrode and the two electrodes, respectively.
Results and discussion
The synthesis mechanism of CHPC composites
The detailed synthesis schematic of CHPC composites is displayed in Fig. 1. Due to the protonation of amino functional groups and the hydrogen bonding between chitosan molecules and solvents, the hydrogen bonding forces between or within the molecules of chitosan are destroyed, which make the chains stretch sufficiently and dissolve in low concentration of acetic acid aqueous solution. After adding gelatin, the charged carboxyl groups (COO−) of gelatin form strong ionic linkage and produce electrostatic interactions with charged amine groups (NH3+) on the chitosan chains. In addition, the polar groups in gelatin (such as –OH, –COOH and –NH2) interact with the solvents and the functional groups in chitosan (–NH2 and –OH), forming hydrogen bonds. The strong intermolecular forces, including electrostatic attraction and hydrogen bond, result in molecule chains of gelatin and chitosan entangling mutually and an interpenetrating or semi-interpenetrating network structure coming into being. Subsequently, the network structure is preserved by freeze-drying process to form sponge carbon precursor in macro-structure, and a large-scale lamellar structure with tangerine pith-like morphology in microscopic scales, as shown in Fig. S1b and S1f. Moreover, the carbonization process not only retains the special tangerine pith morphology, but also effectively develops abundant pores with the help of KHCO3. Accordingly, KHCO3 can be completely decomposed into K2CO3 at 200 °C and the K2CO3 is further pyrolyzed into K2O, as follows (1)–(2) [33]. Moreover, carbon is consumed by the reaction of carbon and H2O with the emission of H2 [Eq. (3)] [37], and the resulting CO2 can be further reduced by carbon to form CO at high temperature [Eq. (4)] [38]. Importantly, the reaction of carbon atoms and the overflow of large quantities of gases lead to the formation of the abundant micropore structure in the chitosan-derived carbon.
The surface and microscopic characterization of carbon materials with different dosage of gelatin and GPC are analyzed by SEM and TEM images in Fig. 2. Compared with that before carbonization (Fig. S1), the morphologies of CHPC series samples (Fig. 2) remain the large-sized lamellas, while the lamellas are broken into smaller sheets due to the activation of KHCO3 during the carbonization process. In sharp contrast, the morphology and structure of GPC change significantly after carbonization, from crisp lamella with tangerine pith-like morphology (Fig. S1d and h) into porous spongy structure (Fig. 2d and h). The difference in morphology before and after carbonization is mainly because the thermal stability of GPC is so poor that the massive porous structure formed easily after carbonization and activation. Thus, the blending of gelatin and chitosan solution not only alters the content of heteroatoms, but also introduces abundant pores into the lamella of chitosan which can dramatically promote the specific surface area. In Fig. 2a, the surface of the carbon sheet is smooth and flat. Interestingly, with the addition of gelatin, the novel tangerine pith-like nanowires form and well scatter on the surface of sheet, as shown in Fig. S1f and S1 g. It is obvious that the nanowires exist stably and become ever more evident after carbonization, as seen in Fig. 2f and g. Moreover, the distribution of the special tangerine pith-like nanowires in CHPC-0.50 (Fig. 2f) is more uniform than that of CHPC-1.0 (Fig. 2g). The difference in distribution is mainly because the aggregation of molecules caused by excessive gelatin. The TEM images of CHPC-0.50 are displayed in Fig. 2i–k. We can clearly observe the nanosheet structure from Fig. 2i, which is consistent with the SEM images. In Fig. 2j and k with larger magnification, lots of densely interconnected nanopores can be seen mainly caused by the activation of KHCO3. The interconnected nanopores not only enlarge the specific surface area of electrode materials for charge storage, but also greatly shorten the transportable distance of electrolyte ion, causing the improvement of capability performance [19, 39].

The illustration for the synthesis of CHPC

SEM images of CPC (a, e), CHPC-0.50 (b, f), CHPC-1.0 (c, g), and GPC (d, h). TEM images of CHPC-0.50 (i–k)
Based on the nanoscale porous characteristics from the SEM and TEM images, the more accurate pore parameters of as-prepared carbon materials are further investigated by N2 adsorption–desorption method and the results are exhibited in Fig. 3 and Table 1. According to the adsorption–desorption isotherm (Fig. 3a), the CHPC series samples have the typical characteristic of type I which has a sharp rise in the adsorption capacity at the low relative pressure (P/P0 < 0.01) and is getting closer to the level at P/P0 > 0.05 [40]. The typical characteristic hints a large number of micropores which can be illustrated by the activation of KHCO3. It is consistent with the result of pore-size distribution (Fig. 3b) which mainly focuses on 0.79–0.89 nm, accompanying significant increasement in the specific surface area of the samples. In the details, the dosage of gelatin also has an obvious influence on the specific surface area and porous structure. Compared with CPC, the specific surface area (SBET) and pore volume (Vpore) of CHPC-0.50 have been greatly increased from 852.12 m2 g−1, 0.36 cm3 g−1 to 927.17 m2 g−1, 0.39 cm3 g−1, mainly in the promotion of micropores. Further, the SBET and Vpore of CHPC-1.0 are further raised to 969.67 m2 g−1, 0.41 cm3 g−1 with the increasement of gelatin dosage. In particular, the rise in number of mesopores is prominent, which leads to the increasement of average pore size with 1.70 nm. Though the average pore size of CHPC-0.50 with 1.67 nm is a little smaller than that of CPC with 1.69 nm and CHPC-1.0 with 1.70 nm, it is larger than that of electrolyte ions (0.36–0.42 nm) [41]. The appropriate average pore size has a positive effect on electrochemical performance and conductivity, especially in the reducing of resistance. The results of N2 adsorption–desorption method confirm the fact that gelatin has been incorporated into composite and effected the porous structures of the samples.

N2 adsorption–desorption isotherms (a) and pore-size distributions (b) of the CPC, CHPC-0.50 and CHPC-1.0 samples
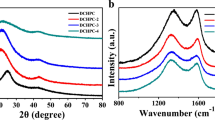
In order to explore the changes of structure characteristics caused by gelatin, the XRD and Raman spectroscopy is adopted and the results are shown in Fig. 4. From all the XRD curves (Fig. 4a), the broad characteristic diffraction peaks located at around 2θ = 24°–25° can be clearly observed, corresponding to the amorphous structure [42]. With the increment of gelatin dosage, the diffraction peaks are flattened, suggesting the presence of high-density micropores [43]. The changes of disordered degree are also confirmed by the Raman spectra, as shown in Fig. 4b. Two characteristic peaks represent the defects in atomic lattices (D-band) and crystallized graphitic carbon (G-band), respectively [44, 45], and the position and the area integral are listed in Table S1. The 2D band centers around 2874 cm−1 are the second order of the D-band, which is the response to the number of graphene layers [46]. As shown in Table S1, with the increase in gelatin dosage, the D and G peaks slightly move toward the shorter wavelength, indicating the increase in disorder degree caused by heteroatom doping. Furthermore, the value of ID/IG (the ratio between the area of the D-band and G-band) is employed to explain the disordered degree of the samples [47]. The CPC has the lowest ID/IG value, meaning the greatest the degree of graphitization among these samples because no gelatin is added. Thus, the ID/IG value is sensitive to the porous structures and the dosage of gelatin. And the values are increased regularly from 3.41 to 3.49 along with the improvement of gelatin content, which is related to more defects generated by the gelatin, and this trend is in good agreement with the analyses of the XRD. These analyses show that the gelatin improves the defects by tuning the porous structures.

XRD patterns (a) and Raman spectra (b) of the CHPC-1.0, CHPC-0.75, CHPC-0.50, CHPC-0.25 and CPC
The changes of element content and bonding configuration caused by the quantity of gelatin are systematically analyzed by XPS measurements, which are shown in Fig. 5. Obviously, three characteristic peaks are observed from the full-scale spectra (Fig. 5a) of the samples, which correspond to C1s (~ 286.1 eV), N1s (~ 400.8 eV) and O1s (~ 530.5 eV) [48]. According to Table 2, the content of nitrogen increases gradually from 5.6 to 5.93 at.% with the addition of gelatin, and the content of oxygen is reduced. And it is noteworthy that the content of heteroatoms (nitrogen and oxygen) of CHPC-0.50 is the highest among the samples. And the existence of heteroatoms helps to improve effectively the specific capacitance by introducing pseudo-capacitance [49]. The high-resolution O1s spectrums (Fig. 5b and Fig. S2a, 2d) display that three peaks at 531.5, 532.4, and 533.4 eV are associated with C=O, C–O, and O=C–O groups [50], and these three groups also can be found in the corresponding C1s spectrum (Fig. S2c) which is decomposed into four peaks corresponding to C–OH (285.3 eV), C=O (286.5 eV), O–C=O (288.8 eV) and C–C (284.7 eV) [51]. The oxygen-containing functional group can increase the wetting capability of carbon electrode in electrolyte and benefit for the ion-penetration diffusion process, fortifying the electrochemical performance [52]. And the N1 s (Fig. 5c and Fig. S2b, 2e) reveals three distinct types of nitrogen, including pyridinic-N (398.5 eV), pyrrolic-N (400.3 eV) and graphitic-N (401.1 eV) [53]. The pyridinic-N and pyrrolic-N impel the carbon electrode to obtain more electrochemical active sites, which contributes to the enhancement of wettability, and the graphitic-N can make positive contribution to the improvement of conductive ability and thermal stability [53, 54]. Deeply, the relative nitrogen contents of each type are shown in Fig. 5d, which are calculated by means of the area of these peaks. Accordingly, the CHPC-0.50 exhibits the high content of graphitic-N (46.32%) which is much higher than that of CPC (31.89%) and CHPC-1.0 (33.09%), while the content of pyridinic-N and pyrrolic-N is slightly lower. With the suitable gelatin dosage, partial pyridinic-N and pyrrolic-N were converted to graphitic-N, leading to the maximum content of graphitic-N in CHPC-0.50. Further increasing the amount of gelatin, the more pyridinic-N and pyrrolic-N come into being that induces the decrease of proportion of the graphitic-N. Thus, graphitic-N helps CHPC-0.50 show great advantages in conductivity and thermal stability and these results can also be verified by TGA curves (Fig. S2f).

Full-scale XPS spectra (a) of the samples, O1s (b) and N1s (c) peaks of CHPC-0.50, and relative nitrogen contents (d) in the samples
Electrochemical performance
The effects of the quantity of gelatin on electrochemical properties are shown in Fig. 6 and Fig. S3. The CV curves (Fig. 6a and Fig. S3a) of the samples show approximate rectangular shape, indicating that electrical double-layer capacitor is predominantly contributed to capacitance. However, wider redox peaks at around − 0.5 to − 0.9 V on the CV curves cannot be ignored, which is related to the pseudo-capacitance provided by heteroatom doping and functional groups [55]. More obviously, the area encircled by the CV curve of CHPC-0.50 reaches the maximum, corresponding to the best capacitance, which is also exemplified by the GCD curve with the longest charge–discharge time (Fig. 6b) and the capacitance performances curves at different current densities (Fig. 6d). The GCD curves (Fig. 6b and Fig. S3b) of all the samples show symmetrical triangular shapes at the current density of 1 A g−1, suggesting good reversible charge–discharge characteristics [56]. Furthermore, the value of specific capacitance can reflect electrochemical performance effectively and visually, which is calculated according to the GCD curves. As displayed in Fig. 6d and Fig. S3d, the specific capacitance of CHPC-0.50 is up to 331 F g−1 at 1 A g−1, which is significantly better than that of CPC (244 F g−1), CHPC-0.25 (301 F g−1), CHPC-0.75 (319 F g−1) and CHPC-1.0 (309 F g−1). And the specific capacitance of CHPC-0.50 can still remain 241 F g−1 even at 10 A g−1, and the capacitance retention can reach up to 73% which is superior to other as-prepared samples, as shown in Tab. S2. The improvement of rate stability is mostly because the layer and porous structure with appropriate pore size provide convenience for the fast and efficient electrolyte ion transportation [57]. The resistances of ion transportation and diffusion are further studied by the EIS test. And the equivalent circuit (Fig. 6c, insert) is used to analyze the EIS curves, which consists of the internal resistance (Rs) of electrolyte and electrode materials, the charge-transfer resistance (Rct) which occurs in the interface of electrode and electrolyte [58, 59], the additional impedance (Zw) caused by concentration and electrochemical polarization at the interface, and the capacitance (C) formed on the surface of electrode [60]. As seen in Fig. 6c and Fig. S3c, a half semicircle at high frequency, corresponding to the Rct, and near vertical linear curve at low frequency are observed. The intercept at the real axis represents the Rs which has little difference for all samples [61]. By contrast, the curve of CHPC-0.50 has smallest semicircle diameter and largest gradient, revealing the optimal capacitive behavior. And the analyses prove again that the layer morphology with hybrid porous structure can provide more storage space and shorten the distance of ion diffusion, and the existence of more heteroatoms can improve the wettability of the electrode materials in electrolyte and introduce the pseudo-capacitance, which have a positive impact on the improvement of electrochemical behavior [62].

Electrochemical capacitive behaviors of the samples. The CV curves (a) at a scan rate of 10 mV s−1, the GCD curves (b) at a current density of 1 A g−1, the Nyquist plots (c), and the dependence of capacitance as a function of current density (d)
The detailed electrochemical performances of CHPC-0.50 are shown in Fig. 7. The shapes of the CV curves (Fig. 7a) transform to spindle with the increasing scan rate, caused by the pseudo-capacitance and the internal resistance which makes it impossible to perform the charge–discharge process in a short time [63]. As shown in Fig. 7b, all the GCD curves at different current densities are almost symmetric, confirming the wonderful capacitive reversibility [64]. The long-term electrochemical stability of CHPC-0.50 is estimated by GCD curves at 10 A g−1 (Fig. 7c). After 10000 cycles, the capacity still remains 90%, demonstrating the excellent cycle stability and the small reduction in capacitance are mainly due to the reduction of diffusion time for ions into the electrode surface. The Ragone plot (Fig. 7d) shows that energy density is about 34 W h kg−1 (with the power density of 900 W kg−1) at 0.5 A g−1, whereas it remains as high as 9 W h kg−1 (with the power density of 18000 W kg−1) at 10 A g−1 for CHPC-0.50, much superior to the most chitosan-based electrode materials [65,66,67,68] and the commercial devices (3–5 W h kg−1) [69].

Electrochemical characterization of the CHPC-0.50 in 6 mol L−1 KOH aqueous electrolyte. a The CV curves at various scan rates, b GCD curves with different current densities, c cyclic stability at a current density of 10 A g−1 over 10000 cycles, d the Ragone plot
Conclusions
In summary, we have successfully fabricated a novel hybrid porous carbon sheets from chitosan as carbon source and gelatin as nitrogen source through facile solution blending. The introduction of gelatin develops a novel tangerine pith-like structure distributed on the surface of carbon sheets and produces abundant interconnected micropores with the help of KHCO3, which greatly increase the specific surface area of the as-prepared carbon materials. Based on high heteroatom content (5.75 at.% of nitrogen and 15.04 at.% of oxygen), specific surface area (927.17 m2 g−1) and appropriate average pore size (1.67 nm), the CHPC-0.5 exhibits excellent electrochemical performances with ideal capacitive performance (331 F g−1 at 1 A g−1), excellent rate capability (241 F g−1 at 10 A g−1) and cycling stability (remains 90% after 10000 cycles). In addition, the energy density can reach 34 W h kg−1 when the power density is 900 W kg−1. The wonderful performances of the obtained carbon materials predict that using renewable and environmentally friendly bio-derived carbon as precursor to produce electrode materials is a potential strategy for the application in energy storage systems.
References
Wu D, Wang Y, Wang F, Wang H, An Y, Gao Z, Fang X, Kai J (2017) Oxygen-incorporated few-layer MoS2 vertically aligned on three-dimensional graphene matrix for enhanced catalytic performances in quantum dot sensitized solar cells. Carbon 123:756–766
Wang J, Ma KY, Zhang J, Liu F, Cheng JP (2017) Template-free synthesis of hierarchical hollow NiSx microspheres for supercapacitor. J Colloid Interface Sci 507:290
Chen A, Yu Y, Xing T, Wang R, Zhang Y, Li Q (2015) Synthesis of graphitic carbon spheres for enhanced supercapacitor performance. J Mater Sci 50:5578–5582. https://doi.org/10.1007/s10853-015-9106-x
Zhang G, Song Y, Hang Z, Jia X, Duan H, Liu J (2016) Nanotube arrays: radially aligned porous carbon nanotube arrays on carbon fibers: a hierarchical 3D carbon nanostructure for high-performance capacitive energy storage. Adv Funct Mater 26:2978
Chen J, Chang B, Liu F, Wei H, Wei W, Lin H, Han S (2019) Modification of porous carbon with nitrogen elements to enhance the capacitance of supercapacitors. J Mater Sci 54:11959–11971. https://doi.org/10.1007/s10853-019-03748-6
Si W, Zhou J, Zhang S, Li S, Xing W, Zhuo S (2013) Tunable N-doped or dual N, S-doped activated hydrothermal carbons derived from human hair and glucose for supercapacitor applications. Electrochim Acta 107:397–405
Zhao X, Gnanaseelan M, Jehnichen D, Simon F, Pionteck J (2019) Green and facile synthesis of polyaniline/tannic acid/rGO composites for supercapacitor purpose. J Mater Sci 54:10809–10824. https://doi.org/10.1007/s10853-019-03654-x
Liu M, Niu J, Zhang Z, Dou M, Wang F (2018) Potassium compound-assistant synthesis of multi-heteroatom doped ultrathin porous carbon nanosheets for high performance supercapacitors. Nano Energy 51:366–372
Huang J, Cao D, Lei T, Yang S, Zhou X, Xu P, Wang G (2013) Structural and electrochemical performance of Al-substituted β-Ni(OH)2 nanosheets electrodes for nickel metal hydride battery. Electrochim Acta 111:713–719
Sarkar A, Chakraborty AK, Bera S, Krishnamurthy S (2018) Novel hydrothermal synthesis of CoS2/MWCNT nanohybrid electrode for supercapacitor: a systematic investigation on the influence of MWCNT. J Phys Chem C 122:18237–18246
Zhang Y, Xuan L, Wang S, Li L, Dou S (2017) Bio-nanotechnology in high-performance supercapacitors. Adv Energy Mater 7:1700592
Wang P, Zhang G, Chen W, Jiao H, Liu L, Wang X, Deng X, Chen Q (2018) Highly porous carbon derived from litchi pericarp for supercapacitors application. J Mater Sci Mater Electron 29:14981–14988
Lei P, Wang Y, Han H, Li X, Liu J, Lu G, Wei T, Wang X et al (2018) 3D self-assembly synthesis of hierarchical porous carbon from petroleum asphalt for supercapacitors. Carbon 134:345–353
Wang RT, Wang PY, Yan XB, Lang JW, Peng C, Xue QJ (2012) Promising porous carbon derived from celtuce leaves with outstanding supercapacitance and CO2 capture performance. ACS Appl Mater Interfaces 4:5800
Chao Z, Zhou X, Cao H, Wang G, Liu Z (2014) Synthesis of porous graphene/activated carbon composite with high packing density and large specific surface area for supercapacitor electrode material. J Power Sour 258:290–296
Chao W, Jiang X, Ding J, Yuan N, Yan P, Zhang R, Liu H (2016) High-performance supercapacitor based on the NaOH activated d-glucose derived carbon. NANO 11:1650075
Hirotomo N, Takashi K (2012) Templated nanocarbons for energy storage. Adv Mater 24:4473–4498
Gao Y, Zhang W, Yue Q, Gao B, Sun Y, Kong J, Zhao P (2014) Simple synthesis of hierarchical porous carbon from Enteromorpha prolifera by a self-template method for supercapacitor electrodes. J Power Sour 270:403–410
Deng J, Xiong TY, Xu F, Li MM, Han CL, Gong YT, Wang HY, Wang Y (2015) Inspired by bread leavening: one-pot synthesis of hierarchically porous carbon for supercapacitors. Green Chem 17:4053–4060
Yunpu Z, Yuqian D, Dongyuan Z, Fulvio PF, Mayes RT, Sheng D (2011) Carbon materials for chemical capacitive energy storage. Adv Mater 23:4828–4850
Zhang Y, Liu L, Zhang L, Yu Y, Lv H, Chen A (2019) Template-free method for fabricating carbon nanotube combined with thin N-doped porous carbon composite for supercapacitor. J Mater Sci 54:6451–6460. https://doi.org/10.1007/s10853-018-03290-x
Fan M, Cheng Y, Tu W, Zhang H (2019) Fabrication of nitrogen-doped hollow carbon nanospheres with variable nitrogen contents using mixed polymer brushes as precursors. J Mater Sci 54:8121–8132. https://doi.org/10.1007/s10853-019-03320-2
Li Z, Li-Zhen F, Meng-Qi Z, Hui G, Suyan Q, Markus A, Maria-Magdalena T (2010) Nitrogen-containing hydrothermal carbons with superior performance in supercapacitors. Adv Mater 22:5202–5206
Sun Q, Li Y, He T (2019) The excellent capacitive capability for N, P-doped carbon microsphere/reduced graphene oxide nanocomposites in H2SO4/KI redox electrolyte. J Mater Sci 54:7665–7678
Ferrero GA, Fuertes AB, Sevilla M (2015) N-doped porous carbon capsules with tunable porosity for high-performance supercapacitors. J Mater Chem A 3:2914–2923
Lee YH, Lee YF, Chang KH, Hu CC (2011) Synthesis of N-doped carbon nanosheets from collagen for electrochemical energy storage/conversion systems. Electrochem Commun 13:50–53
Gao S, Chen Y, Fan H, Wei X, Hu C, Luo H, Qu L (2014) Large scale production of biomass-derived N-doped porous carbon spheres for oxygen reduction and supercapacitors. J Mater Chem A 2:3317–3324
Maria-Magdalena T, White Titirici RJ, Brun N, Budarin VL, Su DS, Del Monte F, Clark JH, Maclachlan M (2014) Sustainable carbon materials. Chem Soc Rev 44:250–290
Liang T, Chen C, Li X, Zhang J (2016) Popcorn-derived porous carbon for energy storage and CO2 capture. Langmuir 32:8042–8049
Biswal M, Banerjee A, Deo M, Ogale S (2013) From dead leaves to high energy density supercapacitors. Energy Environ Sci Technol 6:1249–1259
Raymundo-Piñero E, Cadek M, Béguin F (2009) Tuning carbon materials for supercapacitors by direct pyrolysis of seaweeds. Adv Funct Mater 19:1032–1039
Lota K, Acznik I, Sierczynska A, Lota G (2016) The capacitance properties of activated carbon obtained from chitosan as the electrode material for electrochemical capacitors. Mater Lett 173:72–75
Zhang F, Liu T, Hou G, Kou T, Yue L, Guan R, Li Y (2016) Hierarchically porous carbon foams for electric double layer capacitors. Nano Res 9:2875–2888
Ling Z, Yu C, Fan X, Liu S, Yang J, Zhang M, Wang G, Xiao N et al (2015) Freeze-drying for sustainable synthesis of nitrogen doped porous carbon cryogel with enhanced supercapacitor and lithium ion storage performance. Nanotechnology 26:374003
Chen Z, Kai L, Sheng L, Lu X, Fu J, Zhang X, Zhang C, Gao B (2017) Porous active carbon layer modified graphene for high-performance supercapacitor. Electrochim Acta 237:102–108
Xu B, Hou S, Cao G, Feng W, Yang Y (2012) Sustainable nitrogen-doped porous carbon with high surface areas prepared from gelatin for supercapacitors. J Mater Chem A 22:19088–19093
Yoon S-H, Lim S, Song Y, Ota Y, Qiao W, Tanaka A, Mochida I (2004) KOH activation of carbon nanofibers. Carbon 42:1723–1729
Yanwu Z, Shanthi M, Stoller MD, Ganesh KJ, Weiwei C, Ferreira PJ, Adam P, Wallace RM et al (2011) Carbon-based supercapacitors produced by activation of graphene. Science 332:1537
Fan X, Zhang W, Qiu J (2015) A layered-nanospace-confinement strategy for the synthesis of two-dimensional porous carbon nanosheets for high-rate performance supercapacitors. Adv Energy Mater 5:1401761
Kurosaki F, Koyanaka H, Tsujimoto M, Imamura Y (2008) Shape-controlled multi-porous carbon with hierarchical micro–meso-macro pores synthesized by flash heating of wood biomass. Carbon 46:850–857
Ue M (1994) Mobility and ionic association of lithium and quaternary ammonium salts in propylene carbonate and γ-butyrolactone. J Electrochem Soc 141(12):3336–3342
Mei L, Zhang Y, Yang L, Liu Y, Yao J (2015) Hollow melamine resin-based carbon spheres/graphene composite with excellent performance for supercapacitors. Electrochim Acta 166:310–319
Wang H, Yi H, Zhu C, Wang X, Hong JF (2015) Functionalized highly porous graphitic carbon fibers for high-rate supercapacitive electrodes. Nano Energy 13:658–669
Chao L, Wang D, Zhao J, Song H, Wei C (2017) A continuous carbon nitride polyhedron assembly for high-performance flexible supercapacitors. Adv Funct Mater 27:1606219
Wei H, Chen H, Ning F, Jing C, Lan G, Wei Q, Liu Y, Lin H et al (2017) Excellent electrochemical properties and large CO2 capture of nitrogen-doped activated porous carbon synthesised from waste longan shells. Electrochim Acta 231:403–411
Zhang Y, Liu X, Wang S, Dou SX, Li L (2016) Interconnected honeycomb-like porous carbon derived from plane tree fluff for high performance supercapacitors. J Mater Chem A 4:10869
Yan W, Alsmeyer DC, Mccreery RL (2002) Raman spectroscopy of carbon materials: structural basis of observed spectra. J Mater Chem A 2:557–563
Jutao J, Xiaogang F, Qiao L, Yanru L, Zhiyang W, Kexing N, Junyan Z (2013) Identifying the active site in nitrogen-doped graphene for the VO2 +/VO (+)2 redox reaction. ACS Nano 7:4764
Qian W, Sun F, Xu Y, Qiu L, Liu C, Wang S, Feng Y (2013) Human hair-derived carbon flakes for electrochemical supercapacitors. Energy Environ Sci 7:379–386
Xiang S, Yang X, Lin X, Chang C, Que H, Mei L (2017) Nitrogen and sulfur co-doped polyurethane-based porous carbon materials as supercapacitors exhibit excellent electrochemical performance. J Solid State Electrochem 21:1457–1465
Yi J, Yan Q, Wu CT, Zeng Y, Wu Y, Lu X, Tong Y (2017) Lignocellulose-derived porous phosphorus-doped carbon as advanced electrode for supercapacitors. J Power Sour 351:130–137
Bin DS, Chi ZX, Li Y, Zhang K, Yang X, Sun YG, Piao JY, Cao AM et al (2017) Controlling the compositional chemistry in single nanoparticles for functional hollow carbon nanospheres. J Am Chem Soc 139:13492–13498
Chen P, Yang JJ, Li SS, Wang Z, Xiao TY, Qian YH, Yu SH (2013) Hydrothermal synthesis of macroscopic nitrogen-doped graphene hydrogels for ultrafast supercapacitor. Nano Energy 2:249–256
Fang Y, Gu D, Zou Y, Wu Z, Li F, Che R, Deng Y, Tu B et al (2010) A low-concentration hydrothermal synthesis of biocompatible ordered mesoporous carbon nanospheres with tunable and uniform size. Angewandte Chemie International Editon 49:7987–7991
Feng H, Hang H, Dong H, Yong X, Cai Y, Lei B, Liu Y, Zheng M (2016) Hierarchical structured carbon derived from bagasse wastes: a simple and efficient synthesis route and its improved electrochemical properties for high-performance supercapacitors. J Power Sour 302:164–173
Fan Y, Yang X, Zhu B, Liu PF, Lu HT (2014) Micro-mesoporous carbon spheres derived from carrageenan as electrode material for supercapacitors. J Power Sour 268:584–590
Kondrat S, Pérez CR, Presser V, Gogotsi Y, Kornyshev AA (2012) Effect of pore size and its dispersity on the energy storage in nanoporous supercapacitors. Energy Environ Sci 5:6474–6479
Ullah S, Khan IA, Choucair M, Badshah A, Khan I, Nadeem MA (2015) A novel Cr2O3-carbon composite as a high performance pseudo-capacitor electrode material. Electrochim Acta 171:142–149
Chang C, Yang X, Xiang S, Lin X, Que H, Mei L (2017) Nitrogen and sulfur co-doped glucose-based porous carbon materials with excellent electrochemical performance for supercapacitors. J Electrochem Soc 164:A1601–A1607
Liang Q, Ye L, Xu Q, Huang ZH, Kang F, Yang QH (2015) Graphitic carbon nitride nanosheet-assisted preparation of N-enriched mesoporous carbon nanofibers with improved capacitive performance. Carbon 94:342–348
Xia Y, Zhang W, Xiao Z, Huang H, Zeng H, Chen X, Chen F, Gan Y et al (2012) Biotemplated fabrication of hierarchically porous NiO/C composite from lotus pollen grains for lithium-ion batteries. J Mater Chem 22:9209–9215
Zhao M, Cui X, Chen L, Xu Y, He Z, Yang S, Yi W (2018) An ordered mesoporous carbon nanosphere-encapsulated graphene network with optimized nitrogen doping for enhanced supercapacitor performance. Nanoscale 10:15379–15386
Xie B, Chen Y, Yu M, Shen X, Lei H, Xie T, Zhang Y, Wu Y (2015) Carboxyl-assisted synthesis of nitrogen-doped graphene sheets for supercapacitor applications. Nanoscale Res Lett 10:332
Hui S, Zhu Y, Bo Y, Wang Y, Wu YP, Du J (2016) Template-free fabrication of nitrogen-doped hollow carbon spheres for high-performance supercapacitors based on a scalable homopolymer vesicle. J Mater Chem A 4:12088–12097
Ba Y, Wei P, Pi S, Zhao Y, Mi L (2018) Nitrogen-doped hierarchical porous carbon derived from a chitosan/polyethylene glycol blend for high performance supercapacitors. RSC Adv 8:7072–7079
Zhu L, Feng S, Smith RL, Qi X (2016) High-performance supercapacitor electrode materials from chitosan via hydrothermal carbonization and potassium hydroxide activation. Energy Technol 5:1–10
Li B, Cheng Y, Dong L, Wang Y, Chen J, Huang C, Wei D, Feng Y et al (2017) Nitrogen doped and hierarchically porous carbons derived from chitosan hydrogel via rapid microwave carbonization for high-performance supercapacitors. Carbon 122:592–603
Huang J, Liang Y, Hang H, Liu S, Cai Y, Dong H, Zheng M, Yong X et al (2017) Ultrahigh-surface-area hierarchical porous carbon from chitosan: acetic acid mediated efficient synthesis and its application in superior supercapacitor. J Mater Chem A 5:24775–24781
Hou J, Cao C, Idrees F, Ma X (2015) Hierarchical porous nitrogen-doped carbon nanosheets derived from silk for ultrahigh-capacity battery anodes and supercapacitors. ACS Nano 9:2556
Acknowledgements
This study was supported by the International Cooperation Foundation of Qilu University of Technology (QLUTGJHZ2018023) and International Intelligent Foundation of Qilu University of Technology (QLUTGJYZ2018024).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yuan, M., Zhang, Y., Niu, B. et al. Chitosan-derived hybrid porous carbon with the novel tangerine pith-like surface as supercapacitor electrode. J Mater Sci 54, 14456–14468 (2019). https://doi.org/10.1007/s10853-019-03911-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03911-z