
Abstract
Compatibilization of blends by nanoparticles can impart the superior properties of the nanoparticles on the matrix and increase the compatibility of the blends. Manipulation of nanoparticles located at the interface is a premise and challenge to achieve. Poly(ethylene glycol) (PEG) was added to polylactide/poly(propylidene carbonate)/graphene oxide (PLA/PPC/GO) composites to manipulate the distribution of GO. By thermodynamic analysis and phase structure investigations, it was demonstrated that PEG could induce the transfer of GO from the PPC phase to the interface or PLA phase. Rheological investigations suggested that the entanglement density between the macromolecular chain and GO was improved, which enhanced the interactions of the macromolecular matrix and nanoparticles. Consequently, the compatibility and mechanical properties were improved significantly. In particular for the samples of PLA/PPC/GO 70/30/0.5 with 2 wt% PEG, the elongation at break was improved by a factor of 10 and the tensile strength was maintained at approximately 45 MPa. This simple physical method is expected to popularize the compatible role of GO in various industrial fields.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biodegradable polyesters have been deemed as the most promising polymers to overcome the increasing environmental pollution problems brought about by nonbiodegradable materials. Polylactide (PLA) and poly(propylidene carbonate) (PPC) are two kinds of representative biodegradable polyesters that are expected to be applied in industrialization because of their good mechanical and processing properties, comparable with those of polyolefin. However, PLA is very brittle with high tensile strength [1], while PPC has excellent tensile toughness with low strength [2]. Many works have reported blending PLA and PPC with different weight ratios to improve the toughness of PLA or enhance PPC [2, 3]. Unfortunately, the poor compatibility of the two components is a great obstacle to obtain the ideal material [2, 4]. To improve their compatibility, introductions of copolymers (block, graft, or star) or reactive modifiers, including poly(vinyl acetate) (PVAc), dicumyl peroxide (DCP), or maleic anhydride (MA), are common methods [2,3,4,5,6].
In the last decade, nanoparticles, including carbon nanotubes, montmorillonite, or graphene oxide (GO), have been reported to improve the compatibility of polymer blends, when they are selectively located at the interface [7,8,9]. Compared with the traditional compatibilization methods, nanoparticles can impart their superior properties on the matrix as well as increase the compatibility of the blends. To achieve the target of increasing compatibility, the manipulation of the location of nanoparticles to the interface is the premise. In theory, the locations of nanoparticles in blends are possibly influenced by thermodynamics, kinetics, and even the surface properties of particles [10]. The chemical modification of nanoparticles is the most common method to manipulate their locations because it can alter the surface properties of nanoparticles and guarantees the uniform dispersion of nanoparticles in blends. For example, we prepared functional GO by grafting PLA to improve the compatibility of PLA/PPC blends and obtained blends with satisfactory toughness and strength [11].
Although the chemical modification of nanoparticles is an effective method to manipulate their distributions and dispersions, its expensive cost continues to hinder its industrial applications. Many efforts have been devoted to seek a physical approach to achieve this goal. In Xu’s work, GO was anchored on starch granules by hydrogen bonding, which avoided reaggregation and significantly strengthened the interfacial interactions of PLA and starch composites [12]. Poly(ethylene glycol) (PEG) has also been reported to form special physical interactions with GO, which promoted the dispersion of GO in PLA homopolymer [13]. Scaffaro prepared a GO-PEG masterbatch from an aqueous solution and used it to melt with PLA. It was found that GO was dispersed in the matrix without aggregation and acted as a compatibilizer for PLA/PEG blends [14].
In this work, we designed a physical method by virtue of PEG to simultaneously control the distributions and dispersions of GO in the blends. In our previous work, GO was found to be preferentially located at the PPC phase in PLA/PPC/GO composites [11]. Because PLA and the low composition of PEG (≤ 50 wt%) are completely miscible in thermodynamics [15,16,17], the special interactions between PEG and GO might cause the changes of the GO distribution in the PLA and PPC matrix. Albeit some works have found that PLA/PEG blends are prone to segregation after a certain time interval, depending on the PEG molecular weight, temperature, and humidity [16, 17], this problem can be neglected if an extremely low content of PEG is loaded into the PLA/PPC/GO composites. Based on this, we added a low concentration of PEG (not more than 2 wt%) into the PLA/PPC/GO composites by solution blending to study the structural and property changes. The investigations of the phase structures and mechanical properties demonstrate that PEG modified both the dispersion and distribution of GO nanoparticles in PLA/PPC blends, which realized significant improvements of the mechanical properties. This work is expected to widen the range of industrial applications of nanoparticles in the compatibilization of blends.
Experimental
Materials
PLA (Natureworks 2003D) with Mw = 200,000 and Mn = 120,000 was purchased. PPC with Mw = 250,000 and Mn = 78,000 was supplied by Changchun Institute of Applied Chemistry, Chinese Academy of Science. PEG with Mw = 20,000 was purchased from Shanpu Chemical Engineering Co. Ltd., Shanghai, China. GO was prepared using the modified Hummers’ method [18, 19], and its characterization results were published in our previous reports [20]. Tetrahydrofuran (THF) and hexamethylene were purchased from Shanghai Reagents Co. Ltd.
Preparation of PLA/PPC/GO composites with different PEG compositions
PLA/PPC/GO composites with different PEG compositions were prepared by the solution blending method. PLA, PPC, and PEG, with a certain ratio, were dissolved in THF to form a PLA/PPC/PEG blend solution. GO was dispersed in THF by ultrasonication for 90 min at 25 °C. Then, the GO suspension and the PLA/PPC/PEG blend solution were mixed together, and underwent magnetic stirring until they formed a homogenous mixture. This mixture was then poured into the precipitant of hexamethylene. After complete precipitation, the sediment was heated at 40 °C for 48 h, and the resulting material was vacuum dried at 40 °C for 1 week to remove any residual solvent. Finally, the PLA/PPC/GO 70/30/0.5 samples, with PEG compositions of 0 wt%, 0.5 wt%, 1 wt%, and 2 wt%, were prepared.
Characterization of PLA/PPC/PEG/GO composites
A contact angle meter (JC2000D, Zhongchen Digital Technology Apparatus Co. Ltd., Shanghai, China) was used to test the contact angles of PLA, PPC, PEG, and GO. Deionized water (γp = 50.8 dyn cm−1 and γd = 22.5 dyn cm−1) and diiodomethane (γp = 2.3 dyn cm−1 and γd = 48.5 dyn cm−1) were used as the test liquids [21].
The dispersion behavior of GO in the PLA/PPC blends, influenced by the different PEG compositions, was first observed by an optical microscope (OM, Nikon Optiphot-2), equipped with a charge-coupled device digital camera. The composites were placed between two pieces of glass slides (10 mm × 10 mm) and melted on a hot stage at 190 °C for 20 min to observe the morphologies of the composites.
The PLA/PPC/PEG/GO composites with different weight ratios were first cut into ultrathin films at room temperature by a microtome (Leica Ultracutr Me1-057) equipped with a glass knife. Then, a transmission electron microscopy (TEM, Jeol JEM-2100F, Japan) with an acceleration voltage of 200 kV was used to observe the morphology of the composites and evaluate the location of GO in the PLLA/PPC blends.
A Mettler-Toledo differential scanning calorimeter (TGA/DSC1/1600LF, Switzerland) was used to characterize the thermal parameters, including the glass transition temperatures (Tg) and melting temperatures (Tm) of the composites. The samples were first heated to 190 °C, at a heating rate of 50 °C min−1, and held for 3 min to erase the thermal history. After that, the samples were quenched to 20 °C, followed by a second heating to 190 °C at a rate of 10 °C min−1.
The thoroughly dried samples of the PLLA/PPC/GO composites with different PEG compositions were hot-pressed at 190 °C for 5 min with a pressure of 10 MPa. Then, they were quickly transferred onto a hot plate of 25 °C with a pressure of 5 MPa for cooling. Electronic universal tensile strength testing equipment (AI-7000M) was used to investigate the tensile properties at a speed of 10 mm min−1. The average results of six replications were reported.
Scanning electron microscopy (SEM, Nova Nano SEM 450, FEI Co. Ltd.) investigations were performed with an acceleration voltage of 5 kV. The samples were cryogenically broken in liquid nitrogen and were coated with gold in vacuum before observation.
A stress-controlled rheometer (TA DRH2000), with a parallel geometry (diameter of 25 mm), was used to investigate the rheological behavior at 190 °C under a nitrogen atmosphere. The samples were hot-pressed at 190 °C into disks with 25 mm diameter and 0.8 mm thickness before testing. Oscillatory frequency sweeps, covering 0.01–100 rad s−1, were performed under a fixed strain of 1% within the linear viscoelastic range.
Results and discussion
Thermodynamic analysis of GO Dispersion in PLA/PPC, PLA/PEG, and PPC/PEG blends
The selective locations of GO in the different blends of PLA, PPC, and PEG in the equilibrium state can be predicted according to the thermodynamic parameters. The wetting coefficient (ω) is a commonly used parameter to predict the location of a filler in polymer blends. It is calculated by Young’s equation (Eq. 1) [22]:
where γGO-POLYMER1 and γGO-POLYMER2 are the interfacial energies between GO and polymer 1 or polymer 2, respectively, and γPOLYMER1-POLYMER2 is the interfacial energy between polymer 1 and polymer 2. When ω is higher than 1, GO is in the polymer 2 phase, when ω is lower than − 1, it is located in the polymer 1 phase, and when ω is between 1 and − 1, the fillers are preferentially located at the phase interface.
The interfacial energy between GO and the polymers, or polymer 1 and polymer 2, can be calculated by the geometric mean (Eq. 2) or harmonic mean (Eq. 3) [21]:
where γ12 is the interfacial energy between two solids, γ1 or γ2 is the surface energy of a solid, which contains dispersive (\( \gamma_{ 1}^{d} \) or \( \gamma_{ 2}^{d} \)) and polar components (\( \gamma_{ 1}^{p} \) or \( \gamma_{ 2}^{p} \)). The surface energy (γ) can be calculated by Fowkes method [23]:
The contact angle values can be used to calculate the surface energy by virtue of the Owens–Wendt equation [24]:
where γs and γl represent the surface energies of a solid and liquid, respectively, and θ is the contact angle. The γ dl and γ pl parameters of a polar (water) and a nonpolar liquid (diiodomethane) can be obtained according to the literatures [25]. The contact angles of the above two liquids on a solid surface can be measured. Then, by combining Eqs. 4 and 5, the γ ds and γ ps parameters of a solid can be obtained.
Table 1 lists the surface parameters and the interfacial energy values of the samples used in this work. The value of ω calculated by Eq. 1 of GO in the PLA/PPC blends is 7.00, which is higher than 1, indicating that the GO should be selectively located at the PPC phase, while the ω values of GO in the PPC/PEG and PLA/PEG blends are 0.58 and 0.62, respectively. These are both between 1 and − 1, indicating that GO would be distributed at the phase interface in the blends in an equilibrium state. Considering that the PLA/PEG blends are completely compatible, PEG might form one homogenous phase with PLA when it is added into the PLA/PPC blends. Therefore, the thermodynamic selection of GO at the interface of the PEG/PPC phase might cause the distributed changes of GO in the PLA/PPC blends, which can be used to modulate the phase structure and physical properties of the blends.
Effect of PEG on GO distribution in PLA/PPC blends
The samples were first melted at 190 °C for 30 min, to diffuse into phases large enough for observation by OM. Figure 1 shows their morphologies. For the sample without PEG, the irregular black PPC phases (arrow labeled), with sizes of approximately 5–15 µm, were dispersed in the white continuous PLA phase, indicating that GO dispersed in the PPC matrix. For the sample with the PEG composition of 0.5 wt%, some PPC phases retained the irregular morphologies with GO agglomerating, while some other dispersed PPC phases formed circles with GO embracing the boundaries (arrow labeled). When the PEG composition reached 1 wt%, most PPC phases appeared as circles surrounded by black GO. When the PEG composition reached 2 wt%, the PPC phase became white dispersed phase with few surrounding GO (arrow labeled). At this moment, GO formed some aggregations in PLA (circles labeled in Fig. 1d). However, the largest diameter of this kind of GO aggregation was approximately 5 µm, which is smaller than that of the GO aggregations in PLA/PPC/GO 70/30/0.5 without PEG.

Optical microscope images of PLA/PPC/GO 70/30/0.5 samples with different PEG compositions at 190 °C. a 0 PEG, b 0.5% PEG, c 1% PEG and d 2% PEG
TEM was further used to observe the distribution of GO in the PLA/PPC/PEG blends (Fig. 2). Like the OM results, for the sample without PEG, the white PPC phase dispersed in the black PLA phase. GO was completely located at the PPC phase and formed large aggregated regions of approximately 5 µm, as labeled by the blue circles. When the PEG composition was 0.5 wt%, GO was still dispersed in the white PPC phase, while the sizes of the GO aggregations were obviously reduced. Increasing the PEG composition to 1 wt%, accompanying the reduction in the GO aggregations, some GO transferred from the PPC phases to the boundaries of PPC (red arrows labeled). When the PEG composition was 2 wt%, the aggregations formed by the GO dispersed in PPC disappeared absolutely. Some GO was located at the boundaries of the PPC phase (red arrows labeled), while some GO was transferred to the PLA phase completely (blue arrows labeled).

TEM images of PLA/PPC/GO 70/30/0.5 samples with different PEG compositions. a 0 PEG, b 0.5% PEG, c 1% PEG and d 2% PEG
Therefore, both the OM and TEM investigations show the same results that the dispersion and distribution of GO were both affected by the addition of PEG. As the PEG composition increased, GO dispersed more uniformly in the blends. Furthermore, GO preferred to transfer from PPC to the PLA phase with the increase in the PEG composition. When the PEG composition was 1 wt% or 2 wt%, most of the GO was selectively located at the boundaries of the PLA and PPC phases. This is expected to improve the interfacial compatibilization between PLA and PPC and cause changes to the properties.
Changes to the GO distribution in the blends might alter the thermal behavior of the blends. The heating scan curves and the crystallization parameters of the samples with different PEG compositions are shown in Fig. 3 and Table 2. According to the DSC results, Tg of PLA (Tg2) moved toward low-temperature regions with the increase in PEG composition, while that of PPC (Tg1) did not exhibit obvious changes. For example, Tg2 decreased from 58.0 to 54.1 °C with the increase in PEG composition from 0 to 2 wt%. This decrease could be related to the plasticization of PEG on PLA or the increase in compatibility of PLA and PPC. It is noteworthy that melting peaks appeared from the PLA (Tm) when the PEG composition increased to above 1 wt%. By contrast, there was no crystallization of PLA when the PEG composition was lower than 1 wt%. The Xc values were calculated by comparing ΔHm with the value of an infinitely large crystal (ΔHm = 93 J g−1) [26]. Although the crystallinity of the two samples with 1 wt% and 2 wt% PEG increased from 7.1 to 15.8%, both Tm values were approximately 152 °C. The unchanged Tm value indicates that the crystallization was influenced more by the location of GO than by the PEG contents. Because the crystallization rate of PLA is slow, GO is an effective nucleation agent for PLA crystallization [27]. When the PEG content was lower than 1 wt%, the GO that dispersed in the PPC phase could not contribute to the nucleation of PLA. While the PEG composition was higher than 1 wt%, the GO that dispersed in the PLA phase or the boundaries of the PLA phase induced the crystallization of PLA. Therefore, the changes in thermal behavior again suggest that the location of GO in the PLA/PPC blends is altered by the addition of PEG.

Heating scan curves of PLA/PPC/GO 70/30/0.5 samples with different PEG contents after quenching from 190 °C. The heating rate is 10 °C min−1
Mechanical properties and microstructures
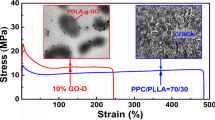
The tensile properties of PLA/PPC/GO 70/30/0.5 with different compositions of PEG are shown in Fig. 4. When the PEG composition was no higher than 2 wt%, the samples exhibited a typical brittle fracture with an elongation at break of less than 15%. When the PEG composition was 2 wt%, the sample broke after yielding, and showed a large elongation at break of approximately 150%, i.e., ten times as long as that of other samples. Simultaneously, the tensile strength increased slightly from 44.4 to 46.2 MPa with the increasing PEG composition from 0 to 2 wt%. However, the tensile modulus changed nonlinearly with the increase in the PEG content. It firstly decreased as the PEG content changed from 0 to 0.5 wt%, then gradually increased as the PEG content increased from 0.5 to 2 wt%. These changes were caused by the common effects of PEG and GO. PEG plays the role of plasticization and results in the increase in the elongation at break and decrease in the tensile strength and tensile modulus. Meanwhile, GO transfers from the PPC phase to the PLA phase or the boundaries, and disperses more uniformly with the increase in the PEG content. These changes might offset the tensile strength and modulus loss caused by PEG. Combining the TEM results, when the PEG composition was 2 wt%, the finely dispersed GO located at the boundaries of PLA/PPC might remedy the tensile strength and tensile modulus loss caused by PEG. Therefore, they both reached the maximum value under the actions of GO enhancing and compatibilization. However, excluding the PLA/PPC samples made compatible by chemical reaction [3, 6], such a large increase in the elongation at break is reported for the first time in physical compatibilization.

aTypical stress–strain curves, b digital photos of the samples after tensile failure and c tensile strength, elongation at break and tensile modulus of PLA/PPC/GO 70/30/0.5 with different composition of PEG
The cryogenically fractured surfaces of the samples were analyzed by SEM to deeply understand the reasons for the mechanical property changes. Figure 5 shows the SEM images of these samples after ultrasonic etching in an acetone/ethanol (1:1) mixed solution for 2 min. For the PLA/PPC/GO composites without PEG (Fig. 5a), a nearly circular PPC phase was dispersed in the PLA phase with a diameter of approximately 15 μm. As the PEG composition increased, the PPC sizes decreased prominently. For the sample with 2 wt% PEG (Fig. 5d), the morphologies of the PPC phase became irregular, and the sizes of most PPC domains decreased to approximately 2–3 µm. This size decrease indicates, again, that the compatibility was increased for the PLA/PPC blends. Combining the OM, TEM, and SEM results, the tensile strength of the composites did not decrease as the PEG composition increased because of the multiple effects including good dispersion of GO, interfacial location of GO, and increased compatibility of PLA and PPC. However, the large increase in the elongation at break is still difficult to understand according to these results.

SEM images of the cryogenically fractured surfaces of PLA/PPC/GO 70/30/0.5 with different PEG compositions after being ultrasonically etched in acetone/ethanol (1:1) mixed solution for 2 min. a 0 PEG, b 0.5% PEG, c 1% PEG and d 2% PEG
To further reveal the reason for the mechanical changes, the rheological behavior was investigated for obtaining detailed structural information. Figure 6a shows the changes of the storage modulus as a function of the angular frequency. With the addition of PEG, the value of G′ showed nonlinear changes. For the sample with 0.5 wt% PEG, G′ was lower than that of the sample without PEG. However, for the sample with 1 wt% PEG, G′ was higher than that of the 0.5 wt% PEG sample. Increasing the PEG composition to 2 wt%, G′ became the highest among all the samples. More importantly, the dependence of G′ on the angular frequency showed a plateau at the low frequency of 0.01–0.1 rad s−1, indicating a transition from “liquid-like” to “solid-like” viscoelastic behavior.

a Storage modulus, G′ and b complex viscosity, |η*|, as functions of angular frequency ω, for PLA/PPC/GO 70/30/0.5 nanocomposites with different PEG compositions
Figure 6b shows the dependence of the complex shear viscosity, |η*|, on the angular frequency. Similar to the changes of G′, the complex viscosity did not linearly decrease with the increase in the PEG content. When the PEG composition was 0.5 wt%, |η*| was lower than that of the samples without PEG. However, |η*| gradually increased with the PEG compositions of 1 wt% and 2 wt%. When the PEG composition was 2 wt%, |η*| became the highest value of all the samples. The nonlinear changes of G′ and |η*| with the composition of PEG suggest that the structural changes have larger effects than the plasticization of PEG on the rheological behavior of the samples.
Figure 7 shows the relationship of the storage modulus G′ and loss modulus G″ of the samples with different PEG compositions, respectively. For the PEG compositions of 0 wt% and 0.5 wt%, there is no cross of G′ and G″, suggesting that the long-range motion of the PLA and PPC chains were very slow. For the samples with PEG compositions of 1 wt% and 2 wt%, the values of G′ and G″ were crossed at the frequencies of 0.019 and 0.20 rad s−1, respectively. This indicates that the addition of PEG played the role of plasticization and improved the chain movement, which shortened the relax time of the chain motion.

Changes of storage modules G′ and loss modules G″ as functions of angular frequency ω for a 0 PEG b 0.5% PEG c 1% PEG and d 2% PEG
Furthermore, the crossover modulus GX (the crossover of G′ and G″) provides information on the average entanglement density (Ve) in the nanocomposites. According to Doi and Edwards’s tube model [28] and de Gennes’ theory [29], the average molar mass of an entanglement strand, Me, and Ve can be obtained by:
where ρ is the amorphous mass density, and G 0 N is the plateau modulus, which can be determined by Eq. (8) according to Ref. [30, 31]:
In this work, the PEG composition was very little (< 2 wt%), and its effect on Mw and Mn can be neglected. Therefore, Mw and Mn can be calculated according to [32]:
where w1 and w2 are the weight fractions of PLA and PPC, respectively. The entanglement density values for the samples with PEG compositions of 1 wt% and 2 wt%, calculated by combining the above equations (Eqs. (6–10)), are 2.8 × 10−2 mol m−3 and 1.49 × 10−1 mol m−3, respectively. The increase in the entanglement density with the increasing PEG content suggests, again, that the microstructures of the composites were modified.
Figure 8 illustrates the changes of the PLLA/PPC/GO nanocomposite structures before and after PEG addition. For the PLLA/PPC/GO nanocomposites without PEG, the PPC phase with large sizes dispersed in the PLA continuous phase because of the poor compatibility of PLA and PPC, and GO aggregated in the PPC phase. After adding PEG, the size of the PPC phase decreased significantly, and the dispersion of GO improved. There are two reasons for these changes: the first is that PEG is compatible with PLA and partially compatible with PPC, imparting PEG with the role of compatibilizer. However, more than this, PEG induces GO to locate at the interface of PLA/PPC and play the role of nano-compatibilizer. Therefore, GO has more opportunities of touching PLA, PEG, and PPC chain than without PEG. The hydroxyl, carboxyl, and epoxy groups on the surface of GO could interact strongly with the polar macromolecules of PLA and PPC, which impart GO the role of “net-points” and result in the improvement of the entanglement density.

Illustration of PLLA/PPC/GO nanocomposites structures before and after PEG
Therefore, these microstructural changes have notable effects on the rheological behavior and mechanical properties. When the PEG composition was lower than 1 wt%, G′ and |η*| decreased with the addition of PEG because of its plasticization. With the increase in the PEG composition to 1 wt%, the entanglement density of the nanocomposite was improved significantly, resulting in the “solid-like” viscoelastic behavior of G′ and the increased value of |η*|. At the same time, the “net-points” formed between GO and the macromolecular chain enhanced the interactions of the macromolecular matrix and nanoparticles; thus, the stress was transferred more efficiently, which greatly improved the tensile strength, tensile modulus, and elongation at break. Particularly for the samples of PLA/PPC/GO 70/30/0.5 with 2 wt% PEG, the existence of a large amount of “net-points” promoted PPC to exhibit its excellent tensile toughness. Besides this, the plasticization of PEG also supplied the high elongation at break. Therefore, the effect of GO physical compatibilization imparted on the PLA/PPC blends is similar to that of reactive compatibilization.
Conclusion
In summary, we designed PLA and PPC blends with GO located at the interface by the simple physical addition of PEG. Combining the thermodynamic and microstructural analyses of the phase structures, it was demonstrated that adding PEG into the PLA/PPC/GO composites improved the dispersion of GO and led to the transfer of GO from the PPC phase to the interface or PLA phase, which improved the compatibility of the blends. Meanwhile, the changes of GO distribution improved the entanglement of the composites and enhanced the interactions of the macromolecular matrix and nanoparticles. Consequently, the PLA/PPC blends showed a great improvement of the elongation at break while maintaining their tensile strength. Therefore, this work provides a simple and effective method to popularize the compatible role of GO in various industrial fields.
References
Rasal RM, Janorkar AV, Hirt DE (2010) Poly(lactic acid) modifications. Prog Polym Sci 35:338–356
Gao J, Bai H, Zhang Q, Gao Y, Chen L, Fu Q (2012) Effect of homopolymer poly(vinyl acetate) on compatibility and mechanical properties of poly(propylene carbonate)/poly(lactic acid) blends. Express Polym Lett 6:860–870
Hwang SW, Park DH, Kang DH, Lee SB, Shim JK (2016) Reactive compatibilization of poly(l-lactic acid)/poly(propylene carbonate) blends: thermal, thermomechanical, and morphological properties. J Appl Polym Sci 133:43388
Zhou L, Zhao G, Jiang W (2016) Effects of catalytic transesterification and composition on the toughness of poly(lactic acid)/poly(propylene carbonate) blends. Ind Eng Chem Res 55:5565–5573
Yao M, Deng H, Mai F, Wang K (2011) Modification of poly(lactic acid)/poly(propylene carbonate) blends through melt compounding with maleic anhydride. Express Polym Lett 5:937–949
Qin S, Yu C, Chen X, Zhou H, Zhao L (2018) Fully biodegradable poly(lactic acid)/poly(propylene carbonate) shape memory materials with low recovery temperature based on in situ compatibilization by dicumyl peroxide. Chin J Polym Sci 36:783–790
Wang S, Pang S, Pan L, Xu N, Huang H, Li T (2016) Compatibilization of poly(lactic acid)/ethylene-propylene-diene rubber blends by using organic montmorillonite as a compatibilizer. J Appl Polym Sci 133:44192
Wu D, Lin D, Zhang J, Zhou W, Zhang M, Zhang Y, Wang D, Lin B (2011) Selective localization of nanofillers: effect on morphology and crystallization of PLA/PCL blends. Macromol Chem Phys 212:613–626
Wu D, Sun Y, Lin D, Zhou W, Zhang M, Yuan L (2011) Selective localization behavior of carbon nanotubes: effect on transesterification of immiscible polyester blends. Macromol Chem Phys 212:1700–1709
Taguet A, Cassagnau P, Lopez-Cuesta JM (2011) Structuration, selective dispersion and compatibilizing effect of (nano)fillers in polymer blends. Prog Polym Sci 39:1526–1563
Li Q, Qin S, Tian X, Chen X, Chen Y, Niu Y, Zhao L (2018) Enhancement of the interface in poly(l-lactide) and poly(propylidene carbonate) blends by introducing of poly(l-lactide)-grafted graphene oxide to improve mechanical properties. Appl Surf Sci 43:739–749
Xu H, Xie L, Wu D, Hakkarainen M (2016) Immobilized graphene oxide nanosheets as thin but strong nanointerfaces in biocomposites. ACS Sustain Chem Eng 4:2211–2222
Jia S, Yu D, Zhu Y, Wang Z, Chen L, Fu L (2017) Morphology, crystallization and thermal behaviors of PLA-based composites: wonderful effects of hybrid GO/PEG via dynamic impregnating. Polymers 9:528
Scaffaro R, Maio A, Lo Re G, Parisi A, Busacca A (2018) Advanced piezoresistive sensor achieved by amphiphilic nanointerfaces of graphene oxide and biodegradable polymer blends. Compos Sci Technol 156:166–176
Lai WC, Liau WB, Lin TT (2004) The effect of end groups of PEG on the crystallization behaviors of binary crystalline polymer blends PEG/PLA. Polymer 45:3073–3080
Sheth M, Kumar RA, Dave V, Gross RA, Mccarthy SP (1997) Biodegradable polymer blends of poly(lactic acid) and poly (ethylene glycol). J Appl Polym Sci 66:1495–1505
Sungsanit K, Kao N, Bhattacharya SN (2012) Properties of linear poly(lactic acid)/polyethylene glycol blends. Polym Eng Sci 52:108–116
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339
Zhou X, Liu Z (2010) A scalable, solution-phase processing route to graphene oxide and graphene ultralarge sheets. Chem Commun 46:2611–2613
Zhao L, Liu X, Zhang R, He H, Jin T, Zhang J (2014) Unique morphology in polylactide/graphene oxide nanocomposites. J Macromol Sci B 54:45–57
Dalal EN (1987) Calculation of solid surface tensions. Langmuir 3:1009–1015
Sumita M, Sakata K, Asai S, Miyasaka K, Nakagawa H (1991) Dispersion of fillers and the electrical conductivity of polymer blends filled with carbon black. Polym Bull 25:265–271
Fowkes FM (1962) Determination of interfacial tensions, contact angles, and dispersion forces in surfaces by assuming additivity of intermolecular interactions in surfaces. J Phys Chem 66:382
Owens DK, Wendt RC (1969) Estimation of the surface free energy of polymers. J Appl Polym Sci 13:1741–1747
Van Oss CJ, Good RJ, Chaudhury MK (1988) Additive and nonadditive surface tension components and the interpretation of contact angles. Langmuir 4:884–891
Fisher E, Sterzel WHJ, Wegner G, Kolloid ZZ (1973) Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reaction. Polymer 251:980–990
Zhao L, Li Q, Zhang R, Tian X, Liu L (2016) Effects of functionalized graphenes on the isothermal crystallization of poly(l-lactide) nanocomposites. Chin J Polym Sci 34:111–121
Doi M, Edwards SF (1988) The theory of polymer dynamics. Oxford University Press, New York
de Gennes PG (1979) Scaling concepts in polymer physics. Cornell University Press, London
Wu S (1989) Chain structure and entanglement. J Polym Sci Part B Polym Phys 27:723–741
Nobile MR, Cocchini F (2001) Evaluation of molecular weight distribution from dynamic moduli. Rheol Acta 40:111–119
Wu S (1987) Entanglement between dissimilar chains in compatible polymer blends: poly(methyl methacrylate) and poly(vinylidene fluoride). J Polym Sci Part B Polym Phys 25:557–566
Acknowledgements
This work was supported by the National Natural Science Foundation, China (Grant No. 51503117), Scientific Research Foundation of Shandong University of Science and Technology for Recruited Talents (2014RCJJ002) and Shandong University of Science and Technology Postgraduate Technology Innovation project SDKDYC180331.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tian, X., Hu, Y., Zhang, J. et al. Blends of poly(l-lactide), poly(propylidene carbonate) and graphene oxide compatibilized with poly(ethylene glycol), and their mechanical properties. J Mater Sci 54, 14975–14985 (2019). https://doi.org/10.1007/s10853-019-03905-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03905-x