
Abstract
In the present research, zeolitic imidazolate framework-67 (ZIF-67) was synthesized through a microwave-assisted method and used as an electrode modifier for simultaneous determination of dopamine (DPM) and paracetamol (PRA). The as-prepared materials were characterized by X-ray diffractometry, scanning electron microscopy, nitrogen adsorption/desorption isotherms, and X-ray photoelectron spectroscopy. It was found that the proposed microwave-assisted approach required a short synthesis time and provided the ZIF-67 with excellent textural properties and high yield rate as compared to conventional hydrothermal processes. The as-synthesized ZIF-67 possessed excellent textural properties and manifested a superior electrocatalytic activity toward the oxidation of PRA and DPM. The electrochemical oxidation of DPM and PRA was investigated by both cyclic voltammetry and differential pulse voltammetry. The ZIF-67 modified electrode caused electrocatalytic oxidation of DPM and PRA in a linear response range from 2.0 × 10−6 to 22 × 10−6 M with the detection limits of 1.3 × 10−6 M and 1.4 × 10−6 M, respectively. A proposed method was successfully applied to analyze DMP and PRA in pharmaceutical preparations.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dopamine (DPM) is an important neurotransmitter involved in human metabolism [1, 2]. It plays an important role in the human brain, and a loss of DPM-containing neurons may result in some serious diseases [3]. Paracetamol (PRA) or acetaminophen is widely used as a pharmaceutical pain reliever [4, 5]. The usage of DPM and PRA above the recommended therapeutic levels can cause adverse side effects. Low PRA concentrations significantly restrict DPM-neurodegeneration, whereas high PRA concentrations do not preserve 6-hydroxydopamine-induced DPM-neurodegeneration [6, 7]. Hence, the simultaneous determination of DPM and PRA is extremely important from both scientific and therapeutic perspectives. Several techniques including spectrophotometry [8, 9], high-performance liquid chromatography (HPLC) [10, 11], spectro-fluorimetry [12], mass spectrometry [13], chemiluminescence [14], and capillary electrophoresis [15] are often used for simultaneous or individual determination of DPM and PRA; however, these techniques are costly and very time-consuming. On contrary, simplicity, low cost, high sensitivity, and rapidness are the main advantages of electrochemical methods [16].
The development of new electrodes and their applications in stripping voltammetric method for the determination of organic substances (DPM, PRA, ascorbic acid, uric acid) have received considerable scientific interests. The critical problem during electrochemical detection of DPM is the interference from other redox active species of similar potential to that required to oxidize or reduce the target molecule, thus the overlapping of voltammetric signals often occurs for a mixture of analytes. This phenomenon could be improved by the use of modified electrodes made of different functional materials, which possess abundant adsorptive sites and manifest excellent catalytic properties with minimal surface fouling effect. In this regard, multi-walled carbon nanotube-supported cobalt nanoparticle-modified electrodes [17], Cu- and Cu/Cu2O nanoparticle-modified electrodes [18], thin pyrolytic carbon films [19], and zirconium nanoparticles decorated reduced graphene oxide-modified electrode [3] have been already employed in stripping voltammetric method.
Modified electrode materials possess several advantageous properties including high conductivity, large surface area, and ordered porous structure with many adsorptive sites. Therefore, porous materials, such as ordered mesoporous materials (MCM–14, SBA–15), nanometal oxides, metal–organic frameworks (MOFs), zeolite, seem to be the perfect choice to develop modified electrodes for the simultaneous determination of DPM and PRA. Zeolitic imidazolate frameworks (ZIFs), a subclass of MOFs, are formed by bridging tetrahedral metal ions (Zn, Co) with imidazolate (IM) [20] and manifest exceptional chemical and thermal stability [21,22,23,24]. ZIF-67 can be formed by bridging 2-methylimidazolate anions (MIM) with cobalt cations [25]. Generally, ZIF crystals are prepared by solvothermal methods in organic liquids (methanol, N, N-dimethylformamide (DMF), and N, N-diethylformamide (DEF)) at high temperatures (> 100 °C). Qian et al. [26] reported the hydrothermal synthesis of ZIF-67 nanocrystals. Pan et al. [27] proposed a novel synthesis method to prepare ZIF–8 (Zn (MIM)2) in aqueous solutions at room temperature. Beldon et al. [28] advocated a ball milling route for ZIF synthesis at room temperature, and ZIF–8 and ZIF-67 were fabricated by a mixed base of ammonium hydroxide and triethylamine.
Although there are many proposals to improve the quality of ZIFs as mentioned above, finding approach for high performance and morphological control has also been taken up by many scientists. The microwave method, due to its high efficiency and short synthesis time, is widely used to fabricate different types of MOFs. MOF–199 synthesized by microwave method exhibits remarkable surface properties [29, 30]. Silva et al. [31] reported the microwave synthesis of Zn-trimesate with a unique cauliflower-like structure. The processing of ZIF–8 and ZIF-67 under microwave irradiation has been also reported [32,33,34]. So far, there are few papers using microwave to synthesize ZIF-67 [35].
ZIF-67 manifests tunable surface functionality, high surface area, and structural flexibility; therefore, it could be a promising candidate for adsorption and separation [36,37,38], electrocatalysis [39], gas storage [36, 40], and drug delivery [41]. However, ZIFs have been rarely utilized for sensing applications. Zhao et al. [42] used ZIF-67 as an electrocatalyst for sensing glutathione. Chen et al. [43] employed ZIF-67 as a potential formaldehyde gas sensor at low temperatures. Wu et al. [44] expounded the usage of ZIF-67 for the preparation of nanocobalt oxide materials for electrochemical applications. However, very few studies have employed ZIF-67 as an electrode material for stripping voltammetric technique [42].
In the present paper, the microwave-assisted synthesis of ZIF-67 was demonstrated and the simultaneous determination of DPM and PRA in stripping voltammetric method using a ZIF-67-modified glassy carbon electrode (GCE) was addressed.
Experimental procedure
Materials
2-Methylimidazole (CH3C3H2N2H, 99%) (denoted as MIM) and cobalt nitrate hexahydrate (Co (NO3)2.6H2O, 99%) were procured from Merck, Germany. Dimethylformamide (DMF; C3H7NO), ethanol (EtOH; C2H5OH), and acetone (ACT; C3H6O, 99% purity) were supplied by HiMedia, India. Paracetamol (C8H9NO2, 99%) (denoted as PRA) and dopamine or 2-(3, 4-dihydroxyphenyl) ethylamine (C8H11NO2, 98%) (denoted as DPM) were purchased from Merck, Germany.
Stock solutions (1000 mg mL−1) of DPM and PRA were prepared by dissolving 0.1000 g of PAR or DPM in deionized water, and then, the working solutions were made by diluting with the same solvent to 100 mL in a calibrated flask. 0.5 M Britton–Robinson buffer solution (B-RBS) was prepared from o-boric (H3BO3), o-phosphoric acid (H3PO4), and acetic acid (CH3COOH). The B-RBS of pH 3–9 was obtained from 0.5 M B-RBS with 0.1 M KOH or 0.1 M H3PO4.
Apparatus
Powder X-ray diffraction patterns (XRD) of ZIF-67 were recorded by a D8 Advance Bruker diffractometer equipped with a Cu-Kα radiation source (λ = 1.5406 Å). A scanning electron microscope (SEM; Hitachi S–4800) was employed to examine surface morphologies of the as-prepared materials. Nitrogen adsorption/desorption isotherms were recorded by a Micromeritics-ASAP 2020 instrument, and the sample was degassed at 120 °C for 5 h. Moreover, the specific surface area was calculated by the Brunauer–Emmett–Teller (BET) model, and the mesoporous surface area was measured by the t-plot method. Voltammetric measurements were performed by a CPA-HH5 computerized polarography analyzer (Vietnam) in which a conventional three-electrode system (consisted of Ag/AgCl/saturated KCl as the reference electrode, a platinum wire as the auxiliary electrode, and a bare GCE (2.8 mm in diameter) or ZIF-67-modified electrode as the working electrode) was used for all electrochemical experiments.
The comparison was carried out according to the modified American Pharmacopeia recommendations (USP XXXII) [45]. The chromatographic determinations were performed in a Schimadzu 2030 HPLC system. The chromatographic conditions were HiQ sil C18 (250 mm × 4.6 mm), detector wavelength 225 nm, mobile phase as water/methanol/acetic acid = 69: 28: 3, and room temperature.
Microwave-assisted synthesis of ZIF-67
An exact amount of MIM (16 mmol) and Co (NO3)2.6H2O (4 mmol) was first dissolved in 200 mL mixture of ethanol and methanol (1:1 in v/v) and then stirred magnetically for 30 min. The resultant mixture was loaded onto a 250-mL Erlenmeyer flask and placed in a microwave system (Fig. 1).

Homemade microwave-assisted synthesis device
After the desired irradiation time, the produced solid was collected by centrifugation, washed with methanol three times, and finally, dried at 120 °C for 24 h, and ZIF-67 synthesized by this process was termed as MW-ZIF-67. ZIF-67 samples were also prepared by the conventional hydrothermal process in order to compare their properties with those of MW-ZIF-67. The same amount of reactants (as in MW-ZIF-67 synthesis) was kept in a Teflon autoclave and placed in an oven at 100 °C or 25 °C (room temperature) for certain times. The produced solid was collected by centrifugation, washed with MeOH three times, and dried at 120 °C for 24 h, and the obtained ZIF-67 samples were denoted as HT-ZIF-67 or RT-ZIF-67, respectively.
Further, product yield was calculated using the following equation:
where mp is the actual amount of ZIF-67, mt is the theoretical amount of ZIF-67 calculated based on cobalt using the chemical reaction of Co(II) + 2MIM → Co(MIM)2.
Voltammetric procedure
Preparation of electrode
The bare electrode was a glass carbon electrode (GCE) with a diameter of 2.8 ± 0.1 mm. GCE was polished with Al2O3 powder (particle size of 0.05 μm), soaked in 2 M HNO3 solution, washed with ethanol and distilled water, and finally, dried naturally at room temperature. The modification of the GCE electrode was carried out according to the following process: ZIF-67 dispersion (2.0 mg/mL) was placed under ultrasonic irradiation for 1 h to obtain solution A. Subsequently, 2.5 μL of solution A was dripped onto the GCE electrode surface, and the solvent was allowed to evaporate at room temperature for 2–3 min. The obtained ZIF-67 modified GCE was denoted as ZIF-67/GCE and stored at room temperature in a desiccator (as-prepared ZIF-67 electrode). The electrochemical treatments of the modified electrode were performed in 0.1 NaOH solution through potential cycling between 0.2 and –1.2 V with a scan rate of 0.2 V s−1 (for ten scans) [46].
Electrochemical measurements
Cyclic voltammetric (CV) experiments were conducted in 0.1 M B-R buffer solution (pH of 3–7) at an applied potential range of 0.2–1.2 V with varying scan rates from 0.02 to 0.20 V s−1, and each measurement was repeated four times. Differential pulse voltammetric (DPV) experiments were performed according to the aforesaid procedure. Electrode rotation was kept off for 5 s, and DPV voltammograms were recorded from 0.1 mV to 1.0 V at a scan rate of 0.02 V s−1.
Panadol Extra (Sanofi-Synthelabo), Hapacol Extra (Hau Giang Pharmaceutical Company), Tatanol (Pymepharco company), Dopamine hydrochloride USP 40 mg/mL (Rotexmedica, Germany), Dopamine hydrochloride 4% 40 mg mL−1 (Warsaw Pharmaceutical Works Polfa S.A, Poland), and Dopamine 200 mg/5 mL (Rotexmedica GmbH Atzneimittelwerk) were used as the pharmaceutical samples in the current study. Five tablets of each sample were precisely weighed and finely ground in a mortar. An adequate amount of each powder was weighed and transferred to a 100-mL calibrated flask containing 0.1 M B-RBs pH 3.4. The standard addition method was adopted to analyze the contents of DPM and PRA in the samples. The samples containing dopamine in liquid form were diluted with distilled water at an appropriate ratio without any further treatment. The desired volume (in mL) of the analytical solution was mixed with 0.1 M BRS buffer pH 3.4 to make the total volume to 10 mL, and the analysis was conducted following the DPV method.
Results and discussion
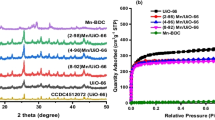
Figure 2 displays XRD patterns of ZIF-67 samples synthesized by means of a microwave-assisted method, where all the diffraction peaks have agreed well with the simulated pattern of ZIF-67 suggesting that the all obtained samples were ZIF-67. The XRD patterns of ZIF-67 synthesized using hydrothermal method at room temperature and at 100 °C were conducted for the sake of comparison (Figure S1). The crystallite size was calculated by means of the Scherrer equation, that is D = Kλ/(β cosθ) where D is crystallite size (nm); K a dimensionless shape factor with a value about 0.9; λ is the X-ray wavelength (0.154 nm); θ diffraction angle; β the full-width at half maximum (FWHM) (radian). The crystallite size for MW-ZIF-67 was found to be larger than those synthesized by the conventional processes (1.622 nm for MW-ZIF-67 (40 min); 1.482 nm for SV-ZIF-67 (1 h) and 1.464 nm for RT-ZIF-67 (3 days)). It implies that the as-prepared ZIF-67 samples possessed very high crystallinity as compared to those processed by conventional methods.

XRD patterns of the MW-ZIF-67 for different microwave irradiation times
Figure 3 exhibits SEM micrographs of ZIF-67 samples synthesized at room temperature for 1, 2, 3, and 9 days. Irregular-shaped particles were observed in the as-synthesized microstructures, and it is evident that particle size started to increase with the increasing aging time. Average particle sizes computed from SEM images were 751, 852, 968 and 1051 nm for 1, 2, 3, and 9 days, respectively (Table 1). Therefore, the diffusion of growth species from the bulk solution resulted in different size distributions in the obtained microstructures.

SEM images of RT-ZIF-67 with different aging times (a: 1 day; b: 2 days; c: 3 days; d: 9 days)
Figure 4 represents SEM images of ZIF-67 samples synthesized by the hydrothermal method at 100 °C for 1, 2, 3, and 4 h. It was found that particle size started to increase with the increasing hydrothermal time and reached ~ 1995 nm (from ~ 835 nm after 1 h) after 4 h reaction (Table 1). At low reaction time, morphologies mainly consisted of both small and large particles; however, with prolonged hydrothermal time, smaller particles were consumed by the Oswald ripening effect to produce larger and uniform particles.

SEM images of HT-ZIF-67 synthesized at different times (a: 1 h; b: 2 h; c: 3 h; d: 4 h)
Figure 5 presents SEM images of ZIF-67 samples processed under different microwave irradiation times. At low irradiation time, morphologies of ZIF-67 crystals tended to be more uniform and regular; however, prolonged irradiation produced irregular-shaped crystals. Larger truncated rhombic dodecahedron crystals and smaller rhombic dodecahedron particles were observed in the as-synthesized morphologies after 20 min microwave irradiation (Fig. 5a). Uniform rhombic dodecahedron particles (average size of ~ 1000 nm) were obtained after 30–40 min irradiation. The rapid diffusion of growth species from the bulk solution resulted in the formation of uniform nanoparticles. Larger particles of about > 1000 nm were produced with prolonged irradiation time (Fig. 5c). Particle size tended to increase with the increasing irradiation time due to the growth of nuclei according to the Oswald ripping effect. The particle sizes of MW-ZIF-67 at 40 min. possessed small standard deviation compared to those of other ZIF-67 indicating the microwave-assisted method provided the particles with high uniform distribution.

SEM images of MW-ZIF-67 synthesized for different microwave irradiation times: a 20 min; b 30 min; c 40 min; d 60 min
The microwave irradiation also promoted the yield rate of ZIF-67. Figure 6 presents the yield rates of ZIF-67 based on cobalt (Co) for different methods. The synthesis of ZIF-67 at room temperature caused a very low yield rate and required long processing time, whereas the hydrothermal treatment at 100 °C enhanced the yield rate. In contrast, microwave irradiation for 40 min manifested a yield rate of 95%, which is similar to that of hydrothermal processing for 4 h.

Yields of as-synthesized products by different methods
Under microwave irradiation, as nucleation occurred in a very short period of time, all nuclei had the same subsequent growth, thus they possessed the same size and formed monosized nanoparticles. Co is one of the most effective electromagnetic wave absorbents; hence, under electromagnetic irradiation, Co ions acted as superheat spots and interacted with imidazolate to form ZIF-67.
Nitrogen adsorption/desorption isotherms were employed to study the textural properties of ZIF-67 (Figure S2). All isotherm curves revealed the typical type-I adsorption isotherms according to IUPAC classification. The significant increase in adsorption volume at a relatively low pressure indicates the existence of micropores in all ZIF-67 samples. In addition, the increase in adsorbed N2 amount near p/p0 = 1 caused high interparticle porosities among the ZIF-67 particles. Table 1 presents surface areas and micropore volumes and sizes of the synthesized ZIF-67 samples. The specific surface area of ZIF-67 prepared using conventional methods were similar to those previously reported (SBET = 316–1319 m2g−1) [47]. It was worth noting that ZIF-67 prepared by means of microwave-assisted method possessed a very large surface area and microvolume (0.98 cm3 g−1; 1935 m2g−1).
CV experiments were performed to compare the electrochemical behavior between bare GCE and ZIF-67-modified GCE (Figure S3). The current response on bare GCE or as-prepared ZIF-67/CGE exhibited broad peaks; especially, the DPM peak was unclear. However, the current response on the ZIF-67/GCE with electrochemical treatment in NaOH solution using CV manifested defined peaks, and the peak–peak separation was found to be 0.19 V. The intensities of the anodic current peak (denoted as Ip) for DPM and PRA on ZIF-67/GCE were, respectively, 4.0- and 1.8-folds higher as compared to bare GCE.
The coverage of ZIF-67 on GCE was investigated by the standard electron transfer redox probe: 1.0 mM K4Fe (CN)6 in 0.1 M KCl (Figure S4). As compared to bare GCE, significant enhancements in both voltammetric peak height and peak separation of ZIF-67/GCE were noticed. For a reversible process, the Randles–Sevcik expression (Eq. 2) was employed to estimate active surface areas of GCE and ZIF-67/GCE [48, 49].
where Ipa represents anodic peak current, n is the number of transferred electrons, A0 is the electrode surface area, D0 is diffusion coefficient, ν signifies scan rate, F is the Faraday constant, R is the universal gas constant, T is temperature, and C0 is the concentration of K4Fe (CN)6. Now, for T = 298 K,
R = 8.314 J mol−1 K−1, and F = 96485.3 C mol−1,
Now, from the slope of the linear plot of Ipaversus v1/2 (using n = 1 and Do = 7.6 × 10−6cm2s−1 [50]), the calculated active areas for GCE and ZIF-67/GCE were found as 0.078 cm2 (0.062 cm2 for geometry area of GCE) and 2.74 cm2, respectively. Therefore, ZIF-67/GCE yielded 35 times larger active surface area as compared to bare GCE (44 times of geometry surface area).
Effects of solvents and ZIF-67 amount
Four common solvents, dimethyl formaldehyde (DMF), methanol (MtOH), ethanol (EtOH), and distilled water (WAT), were used in the current experiment, and the corresponding CV results are presented in Figure S5a. All four solvents manifested well-defined peaks for both DPM and PRA. DMF and WAT yielded shaper peaks and higher intensities among all solvents; however, as WAT is more volatile than DMF, it was selected as the main solvent to disperse ZIF-67.
The effects of ZIF-67 suspension volume (in the range of 2.5–15 μL (2 mg mL−1)) on GCE were also investigated (Figure S5b). It is evident that the intensities of Ip for both PRA and DPM increased with the increasing suspension volume, thus causing an increase in electrode surface porosity due to the enhancement in the effective surface area of the modified electrode. However, for suspension volume greater than 7.5 μL, as the larger film thickness of ZIF-67/GCE caused considerable resistance against electron transfer, a decrease in Ip was noticed.
Effects of pH
The effects of pH (in the range of 2.5~5.3) on voltammetric responses of DPM and PRA were investigated using the Britton–Robinson buffer solution (Fig. 7a). It is evident from Fig. 7b that the intensities of Ip for PRA increased with the increasing pH in the range of 2.5–3.4 and then started to change irregularly, whereas Ip for DPM increased with the increasing pH from 2.5 to 4 and decreased slightly afterward. Therefore, the pH value of about 3.4 was selected for subsequent analytical experiments.

a CV curves; b change of peak currents, and c linear plots of peak potential versus pH for DPM and PRA at different pH values
Within the pH range of 2.5–5.3, the oxidation peak potentials (Ep, PRA and Ep, DPM) were found to be dependent on pH. As can be seen from Fig. 10c, the shifting potentials for DPM and PRA toward less positive values as the pH gradually increased suggested participating protons in the oxidation reactions. Hence, the linear regression equations can be expressed as follows:
The values of 0.0559 V/pH for DPM and 0.0479 V/pH for PRA were close to the Nernstian slope of 0.059 V/pH, thus it implies that an equal number of transferred electrons and hydrogen ions took part in the electrode reaction.
Effects of scan rate
The effects of ν on both Ep and Ip are depicted in Fig. 9a. When the electro-oxidation reaction was reversible, Ep was found to be independent on v. It is clear from Fig. 8a that peak potential was slightly changeable with the increasing scan rate; therefore, it can be inferred that electron transfers during DPM and PRA electro-oxidation were quasi-reversible. The linear plots of Ip against v1/2 determine whether DPM and PRA electro-oxidation involved controlled adsorption or diffusion. If the intercept of the plot does not pass through the origin, then it can be assumed that the electro-oxidation process is controlled by adsorption [51, 52].

a Effect of scan rate on Ep and Ip; b The linear plot Ep versus lnv; c The linear plot of Ip, PRA, Ip, DAversus v1/2 (Erange = (–0.2) – (+1.2 V); Eacc = 0; tacc = 0; 0.1 M B-R BS pH 3.4)
The plots of IP,DPM and IP,RA against v1/2 were highly linear (r = 0.992 ~ 0.998 and p < 0.001) (Fig. 8b). The intercepts did not pass through the origin because the 95% confidence interval did not contain a value of 0 (varied from 1.8 to 2.2 for DPM and from 5.0 to 6 for PRA); hence, it expresses that electro-oxidation reactions for both DPM and PRA were controlled by adsorption process.
The Laviron equation [53] was employed to express the relationship between peak potential (Ep) and the natural algorithm of scan rates.
where α is the charge transfer coefficient, ks is the heterogeneous electron transfer rate constant of a surface-confined redox couple, n is the number of transferred electrons, v signifies scan rate (V s−1), E0 is the formal redox potential, T = 298 K, R = 8.314 J mol−1 K−1, and F = 96485.3 C mol−1.
The plots of Ep, PRA and Ep, CAF against lnv are given in Fig. 8c, and the linear regression equations can be formulated as follows:
The αn values for DPM and PRA were estimated as 1.12 and 0.95, respectively, and the value of α was assumed as 0.5, which is generally considered for a totally reversible system [54]. Therefore, the values of n for PRA and DPM electro-oxidation were found as 2.24 and 1.9, respectively. Consequently, assuming n = 2 for PRA and n = 2 for DPM and based on the evidence above and on previous studies [55, 56], an assumption of two protons and two electrons taking part in the oxidation of PAR and DPM was reasonably accepted.
The favorable signal-promoting effect indicates that ZIF-67 enhanced the electron transfer rates of DPM and PRA and manifested excellent electrocatalytic activity toward the redox reaction. ZIF-67 contains Co(II) and imidazole rings of sp2- conjugated bond (π–π interaction). The π–π stacking interaction between phenyl structures of analytes and the three-dimensional imidazolate structure of ZIF-67 facilitated the accumulation of PRA and DPM on the modified electrode surface. In addition, the coordination of nitrogen atoms in PRA and DPM with Co(II) ions in ZIF-67 attracted analytes to the modified electrode surface. Therefore, the combination of these effects expedited the transfer of electrons and resulted in enhanced voltammetric signals. The oxidation mechanisms for PRA and DPM at the modified electrodes are shown in Fig. 9.

A schematic representation for the oxidation of PRA and DPM at ZIF-67/GCE
Effects of operational parameters
The operational parameters of the differential pulse technique were also studied in detail (Figure S6). An accumulation potential (Eacc) of − 0.6 V, an accumulation time (tacc) of 5 s, and a pulse amplitude (ΔE) of 0.06 V were found to be optimal in order to obtain the best voltammetric signal.
Effect of interferents
Some inorganic salts and organic compounds coexist with DPM and PRA in the solution; therefore, it is important to study their effects on the intensities of anodic peak currents. The relative error deviation (RSD) of anodic peak currents was estimated during DPM and PRA oxidation with and without interferents. The interference was studied by the addition of these compounds to a solution containing an equal concentration of 5 × 10−4 mol L−1 DPM and PRA (pH of 3.4) (Table S1-6). Inorganic salts including K2CO3, CaCl2, NaNO3, and (NH4)2SO4 and glucose yielded no effects on anodic peak current as the interference/analyte molar ratio reached 310 with RSD < 5%. However, uric acid manifested profound effects on anodic peak current for DPM and PRA. Either a tenfold higher concentration of uric acid or a 40-fold higher concentration of uric acid had effect on DPM signal or PRA signal with RSD > 5%, respectively.
Repeatability and limit of detection
The repeatability of the modified electrode was confirmed by means of successive measurement of DPV signals (for nine times) at different DPM and PRA concentrations (Figure S7). The values of RSD for CDPM = 6 × 10−4 M, CPRA = 3 × 10−4 M, CDPM = 4 × 10−5 M, CPRA = 2 × 10−5 M, CDPM = 2 × 10−4 M, and CPRA = 10−4 M were estimated as 0.19%, 0.66%, 1.46%, 4.36%, 0.79%, and 3.81%, respectively (these values are all lower than the value of 1/2 RSDHorwitz predicted [57]). Therefore, such acceptable RSD values of successive measurements imply that ZIF-67/GCE can be repeatedly utilized for the detection of DPM and PRA in both low and high concentrations.
The effects of analyte concentrations on anodic peak current were analyzed at optimal conditions of operational parameters. The DPV curves of PRA and DPM were studied in 0.1 M B-R BS pH of 3.4. Figure 10a displays the DPV curves for different DPM concentrations (2.0 × 10−6 to 44 × 10−6 M) (the concentration of PRA was kept constant at 1 × 10−5 M), whereas Fig. 10b presents the DPV curves for different PRA concentrations (2 × 10−6 to 44 × 10−6 M) (the concentration of DPM was kept constant at 1 × 10−5 M). It is discernible that oxidation peak currents increased with the increasing concentrations from 2.0 × 10−6 to 44 × 10−6 M; hence, it indicates that oxidation reactions occurred independently. The linear regressions of Ip,DPM versus CDPM and Ip,PRA versus CPRA can be expressed as follows.

DPV curves of DPM and PRA oxidation a DPM concentration varying from 2.0 × 10−6 to 4.4 × 10−4 M and CPRA = 1 × 10−5 M; b PRA concentration varying from 2.0 × 10−6 to 4.4 × 10−4 M and CPRA = 1 × 10−5 M)
The values of limit of detection (LOD) (signal/noise = 3) for DPM and PRA were calculated as 2 × 10−6 M and 1.0 × 10−6 M, respectively (in the range of 2.0 × 10−6–44 × 10−6 M).
Figure 11a presents the DPV curves for DPM and PRA in the concentration range of 2.0 × 10−6 M to 22 × 10−6 M, and oxidation peaks were found at ~ 0.315 V for DPM and at ~ 0.485 V for PRA. Therefore, the peak separation of ~ 0.170 V allowed the simultaneous determination of DPM and PRA. It was found that peak current linearly depended on concentration (Fig. 11b).

a DPV curves of DPM and PRA oxidation with increasing concentrations of CDPM and CPRA from 2 × 10−6 to 22 × 10−6 M; b Linear plot of Ip,DPM versus CDPM and Ip,PRA versus CPRA
In the linear range of 2 × 10−6 to 22 × 10−6 M, the values of LOD for DPM and PRA were measured as 1.3 × 10−6 M and 1.4 × 10−6 M, respectively. It is worth noting that the LOD of DPM in the presence of PRA was very close to the obtained value (2 × 10−6 M) in the fixing PRA and that of PRA in the present of DPM was also close to the obtained value (1.0 × 10−6 M) in the fixing DPM. It also proves that the electro-oxidation of DPM and PRA on the modified electrode surface occurred independently.
It is evident from Table 2 that the obtained LOD values of DPM and PRA are comparable with the previously reported results [7, 58,59,60,61,62]. Therefore, ZIF-67 could be used as an effective electrode modifier for the determination of DPM and PRA.
Real sample analysis
The proposed method was employed to analyze the contents of DPM and PRA in six different commercial pharmaceutical tablets: Panadol Extra (Sanofi-Synthelabo), Hapacol Extra (Hau Giang Pharmaceutical Company), Tatanol (Pymepharco Company), Dopamine hydrochloride USP 40 mg/mL (Rotexmedica, Germany), Dopamine hydrochloride 4% 40 mg mL−1 (Warsaw Pharmaceutical Works Polfa S.A., Poland), and Dopamine 200 mg/5 mL (Rotexmedica GmbH Atzneimittelwerk).
Table 3 compares the amounts of DPM and PRA obtained from the proposed method and the HPLC method, and the paired sample t test was applied to analyze the difference. With the significant level of α = 0.05, the paired sample t test revealed that the amounts of DPM (t (2) = –0.567, p = 0.628 > 0.05) and PRA (t (2) = 3.351, p = 0.079 > 0.05) determined by the proposed method were found in three pharmaceutical preparations and by the HPLC method was not statistically different. The effects of matrix interference were confirmed by means of recovery values of spiked samples, and it was found that average recoveries ranged from 98 to 101% for DPM and from 99 to 102% for PRA. Therefore, it can be inferred that the proposed method did not suffer from any significant effect of matrix interference.
Conclusion
ZIF-67 was successfully synthesized by the microwave-assisted process. This method provided ZIF-67 with advantages over conventional approaches in terms of short synthetic time, excellent surface properties and high performance. It was used as an electrode modifier to determine DPM and PRA by using DP method. The electrochemistry responses of DPM and PRA at ZIF-67/GCE were significantly enhanced which can be attributed to the high adsorption capacity, excellent electron transfer efficiency. The modified electrode manifests to be promising for DPM and PRA with many desirable properties including high sensitivity, low detection limit, and high reproducibility. The proposed DP method was employed for the determination of DPM and PRA in pharmaceutical samples using standard addition method and the obtained results were found to be satisfactory. The figures of ZIF-67 could make them as perspective materials in electrochemical analysis.
References
Wightman RM, May LJ, Michael AC (1988) Detection of dopamine dynamics in the brain. Anal Chem 60:769A–793A
Dutt VE, Mottola H (1974) Determination of uric acid at the microgram level by a kinetic procedure based on a pseudo-induction period. Anal Chem 46:1777–1781
Vilian AE, Rajkumar M, Chen S-M (2014) In situ electrochemical synthesis of highly loaded zirconium nanoparticles decorated reduced graphene oxide for the selective determination of dopamine and paracetamol in presence of ascorbic acid. Coll Surf B Biointerfaces 115:295–301
Criado A, Cárdenas S, Gallego M, Valcárcel M (2000) Continuous flow spectrophotometric determination of paracetamol in pharmaceuticals following continuous microwave assisted alkaline hydrolysis. Talanta 53:417–423
Rodenas V, Garcıa M, Sanchez-Pedreno C, Albero M (2000) Simultaneous determination of propacetamol and paracetamol by derivative spectrophotometry. Talanta 52:517–523
Locke CJ, Fox SA, Caldwell GA, Caldwell KA (2008) Acetaminophen attenuates dopamine neuron degeneration in animal models of Parkinson’s disease. Neurosci Lett 439:129–133
Kutluay A, Aslanoglu M (2014) An electrochemical sensor prepared by sonochemical one-pot synthesis of multi-walled carbon nanotube-supported cobalt nanoparticles for the simultaneous determination of paracetamol and dopamine. Anal Chim Acta 839:59–66
Mamiński M, Olejniczak M, Chudy M, Dybko A, Brzózka Z (2005) Spectrophotometric determination of dopamine in microliter scale using microfluidic system based on polymeric technology. Anal Chim Acta 540:153–157
Hanaee J (1997) Simultaneous determination of acetaminophen and codeine in pharmaceutical preparations by derivative spectrophotometry. Pharm Acta Helv 72:239–241
Wilson JM, Slattery JT, Forte AJ, Nelson SD (1982) Analysis of acetaminophen metabolites in urine by high-performance liquid chromatography with UV and amperometric detection. J Chromatogr B Biomed Sci Appl 227:453–462
Ravisankar S, Vasudevan M, Gandhimathi M, Suresh B (1998) Reversed-phase HPLC method for the estimation of acetaminophen, ibuprofen and chlorzoxazone in formulations. Talanta 46:1577–1581
Wang HY, Sun Y, Tang B (2002) Study on fluorescence property of dopamine and determination of dopamine by fluorimetry. Talanta 57:899–907
Curtius HC, Wolfensberger M, Steinmann B, Redweik U, Siegfried J (1974) Mass fragmentography of dopamine and 6-hydroxydopamine: application to the determination of dopamine in human brain biopsies from the caudate nucleus. J Chromatogr A 99:529–540
Easwaramoorthy D, Yu Y-C, Huang H-J (2001) Chemiluminescence detection of paracetamol by a luminol-permanganate based reaction. Anal Chim Acta 439:95–100
Zahao S, Xiao D, Bai W, Yuan H (2006) Capillary Electrophoresis with chemiluminescence detection of Paracetamol. Anal Chim Acta 559:195–199
Xiao L, Xu H, Zhou S, Song T, Wang H, Li S, Gan W, Yuan Q (2014) Simultaneous detection of Cd(II) and Pb(II) by differential pulse anodic stripping voltammetry at a nitrogen-doped microporous carbon/Nafion/bismuth-film electrode. Electrochim Acta 143:143–151
Kutluay A, Aslanoglu M (2014) Nickel nanoparticles functionalized multi-walled carbon nanotubes at platinum electrodes for the detection of bromhexine. Sens Actuators B Chem 192:720–724
Devaraj M, Saravanan R, Deivasigamani R, Gupta VK, Gracia F, Jayadevan S (2016) Fabrication of novel shape Cu and Cu/Cu2O nanoparticles modified electrode for the determination of dopamine and paracetamol. J Mol Liq 221:930–941
Keeley GP, McEvoy N, Nolan H, Kumar S, Rezvani E, Holzinger M, Cosnier S, Duesberg GS (2012) Simultaneous electrochemical determination of dopamine and paracetamol based on thin pyrolytic carbon films. Anal Methods 4:2048–2053
Yao J, Wang H (2014) Zeolitic imidazolate framework composite membranes and thin films: synthesis and applications. Chem Soc Rev 43:4470–4493
Park KS, Ni Z, Côté AP, Choi JY, Huang R, Uribe-Romo FJ, Chae HK, O’Keeffe M, Yaghi OM (2006) Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc Natl Acad Sci 103:10186–10191
Li J-R, Kuppler RJ, Zhou H-C (2009) Selective gas adsorption and separation in metal–organic frameworks. Chem Soc Rev 38:1477–1504
Shekhah O, Liu J, Fischer R, Wöll C (2011) MOF thin films: existing and future applications. Chem Soc Rev 40:1081–1106
Tran UP, Le KK, Phan NT (2011) Expanding applications of metal − organic frameworks: zeolite imidazolate framework ZIF-8 as an efficient heterogeneous catalyst for the knoevenagel reaction. Acs Catal 1:120–127
Banerjee R, Phan A, Wang B, Knobler C, Furukawa H, O’keeffe M, Yaghi OM (2008) High-throughput synthesis of zeolitic imidazolate frameworks and application to CO2 capture. Science 319:939–943
Qian J, Sun F, Qin L (2012) Hydrothermal synthesis of zeolitic imidazolate framework-67 (ZIF-67) nanocrystals. Mater Lett 82:220–223
Pan Y, Liu Y, Zeng G, Zhao L, Lai Z (2011) Rapid synthesis of zeolitic imidazolate framework-8 (ZIF-8) nanocrystals in an aqueous system. Chem Commun 47:2071–2073
Beldon PJ, Fábián L, Stein RS, Thirumurugan A, Cheetham AK, Friščić T (2010) Rapid room-temperature synthesis of zeolitic imidazolate frameworks by using mechanochemistry. Angew Chem 122:9834–9837
Minh TT, Phong NH, Van Duc H, Khieu DQ (2018) Microwave synthesis and voltammetric simultaneous determination of paracetamol and caffeine using an MOF-199-based electrode. J Mater Sci 53:2453–2471. https://doi.org/10.1007/s10853-017-1715-0
Seo Y-K, Hundal G, Jang IT, Hwang YK, Jun C-H, Chang J-S (2009) Microwave synthesis of hybrid inorganic–organic materials including porous Cu3 (BTC) 2 from Cu(II)-trimesate mixture. Microporous Mesoporous Mater 119:331–337
da Silva CTP, Safadi BN, Moisés MP, Meneguin JG, Arroyo PA, Fávaro SL, Girotto EM, Radovanovic E, Rinaldi AW (2016) Synthesis of Zn-BTC metal organic framework assisted by a home microwave oven and their unusual morphologies. Mater Lett 182:231–234
Bao Q, Lou Y, Xing T, Chen J (2013) Rapid synthesis of zeolitic imidazolate framework-8 (ZIF-8) in aqueous solution via microwave irradiation. Inorg Chem Commun 37:170–173
Bux H, Liang F, Li Y, Cravillon J, Wiebcke M, Caro Jr (2009) Zeolitic imidazolate framework membrane with molecular sieving properties by microwave-assisted solvothermal synthesis. J Am Chem Soc 131:16000–16001
Yang L, Lu H (2012) Microwave-assisted Ionothermal synthesis and characterization of zeolitic imidazolate framework-8. Chin J Chem 30:1040–1044
Lanchas M, Arcediano S, Aguayo AT, Beobide G, Castillo O, Cepeda J, Vallejo-Sánchez D, Luque A (2014) Two appealing alternatives for MOFs synthesis: solvent-free oven heating versus microwave heating. RSC Adv 4:60409–60412
H-c Guo, Shi F, Z-f Ma, X-q Liu (2010) Molecular simulation for adsorption and separation of CH4/H2 in zeolitic imidazolate frameworks. J Phys Chem C 114:12158–12165
Zhang C, Xiao Y, Liu D, Yang Q, Zhong C (2013) A hybrid zeolitic imidazolate framework membrane by mixed-linker synthesis for efficient CO 2 capture. Chem Commun 49:600–602
Zhong G, Liu D, Zhang J (2018) The application of ZIF-67 and its derivatives: adsorption, separation, electrochemistry and catalysts. J Mater Chem A 6:1887–1899
Yang H, He X-W, Wang F, Kang Y, Zhang J (2012) Doping copper into ZIF-67 for enhancing gas uptake capacity and visible-light-driven photocatalytic degradation of organic dye. J Mater Chem 22:21849–21851
Hu Y, Liu Z, Xu J, Huang Y, Song Y (2013) Evidence of pressure enhanced CO2 storage in ZIF-8 probed by FTIR spectroscopy. J Am Chem Soc 135:9287–9290
Zheng H, Zhang Y, Liu L, Wan W, Guo P, Nyström AM, Zou X (2016) One-pot synthesis of metal–organic frameworks with encapsulated target molecules and their applications for controlled drug delivery. J Am Chem Soc 138:962–968
Zhao J, Wei C, Pang H (2015) Zeolitic imidazolate framework-67 rhombic dodecahedral microcrystals with porous 110 facets as a new electrocatalyst for sensing glutathione. Part Part Syst Charact 32:429–433
Chen E-X, Yang H, Zhang J (2014) Zeolitic imidazolate framework as formaldehyde gas sensor. Inorg Chem 53:5411–5413
Wu R, Qian X, Rui X, Liu H, Yadian B, Zhou K, Wei J, Yan Q, Feng XQ, Long Y (2014) Zeolitic imidazolate framework 67-derived high symmetric porous Co3O4 hollow dodecahedra with highly enhanced lithium storage capability. Small 10:1932–1938
Pharmacopeia US, Revision X (1995) US Pharmacopeial convention. Inc, Rockville, MD 1161–1162
Wang M, Jiang X, Liu J, Guo H, Liu C (2015) Highly sensitive H2O2 sensor based on Co3O4 hollow sphere prepared via a template-free method. Electrochim Acta 182:613–620
Li Y, Zhou K, He M, Yao J (2016) Synthesis of ZIF-8 and ZIF-67 using mixed-base and their dye adsorption. Microporous Mesoporous Mater 234:287–292
Rezaei B, Damiri S (2008) Voltammetric behavior of multi-walled carbon nanotubes modified electrode-hexacyanoferrate (II) electrocatalyst system as a sensor for determination of captopril. Sens Actuators B Chem 134:324–331
Pattar VP, Nandibewoor ST (2015) Electroanalytical method for the determination of 5-fluorouracil using a reduced graphene oxide/chitosan modified sensor. RSC Adv 5:34292–34301
Bagoji AM, Nandibewoor ST (2016) Electrocatalytic redox behavior of graphene films towards acebutolol hydrochloride determination in real samples. New J Chem 40:3763–3772
Soleymani J, Hasanzadeh M, Shadjou N, Jafari MK, Gharamaleki JV, Yadollahi M, Jouyban A (2016) A new kinetic–mechanistic approach to elucidate electrooxidation of doxorubicin hydrochloride in unprocessed human fluids using magnetic graphene based nanocomposite modified glassy carbon electrode. Mater Sci Eng, C 61:638–650
Bard AJ, Faulkner LR (2001) Fundamentals and applications. Electrochem Methods 2:482
Laviron E (1979) General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanal Chem Interfacial Electrochem 101:19–28
Li C (2007) Electrochemical determination of dipyridamole at a carbon paste electrode using cetyltrimethyl ammonium bromide as enhancing element. Colloids Surf B 55:77–83
Zen J-M, Ting Y-S (1997) Simultaneous determination of caffeine and acetaminophen in drug formulations by square-wave voltammetry using a chemically modified electrode. Anal Chim Acta 342:175–180
Sanghavi BJ, Srivastava AK (2010) Simultaneous voltammetric determination of acetaminophen, aspirin and caffeine using an in situ surfactant-modified multiwalled carbon nanotube paste electrode. Electrochim Acta 55:8638–8648
Horwitz W, Albert R (1997) Quality issuesthe concept of uncertainty as applied to chemical measurements. Analyst 122:615–617
Kumar SA, Tang C-F, Chen S-M (2008) Electroanalytical determination of acetaminophen using nano-TiO2/polymer coated electrode in the presence of dopamine. Talanta 76:997–1005
Li M, Jing L (2007) Electrochemical behavior of acetaminophen and its detection on the PANI–MWCNTs composite modified electrode. Electrochim Acta 52:3250–3257
Liu S-Q, Sun W-H, Hu F-T (2012) Graphene nano sheet-fabricated electrochemical sensor for the determination of dopamine in the presence of ascorbic acid using cetyltrimethylammonium bromide as the discriminating agent. Sens Actuators B Chem 173:497–504
Alipour E, Majidi MR, Saadatirad A, mahdi Golabi S, Alizadeh AM (2013) Simultaneous determination of dopamine and uric acid in biological samples on the pretreated pencil graphite electrode. Electrochim Acta 91:36–42
Cheemalapati S, Palanisamy S, Mani V, Chen S-M (2013) Simultaneous electrochemical determination of dopamine and paracetamol on multiwalled carbon nanotubes/graphene oxide nanocomposite-modified glassy carbon electrode. Talanta 117:297–304
Manjunatha R, Nagaraju DH, Suresh GS, Melo JS, D’Souza SF, Venkatesha TV (2011) Electrochemical detection of acetaminophen on the functionalized MWCNTs modified electrode using layer-by-layer technique. Electrochim Acta 56:6619–6627
Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under Grant Number 104.06-2018.15.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tu, N.T.T., Sy, P.C., Thien, T.V. et al. Microwave-assisted synthesis and simultaneous electrochemical determination of dopamine and paracetamol using ZIF-67-modified electrode. J Mater Sci 54, 11654–11670 (2019). https://doi.org/10.1007/s10853-019-03709-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03709-z