
Abstract
Humic acid (HA), which contains abundant carboxyl groups and hydroxyl groups, is one of the major constituents of dissolved organic matter. The increase of HA in natural waters worldwide has caused great trouble in water treatment and water health. Photocatalysis is a promising technology for degrading HA. In this study, graphene oxide, TiO2 and different amounts of urea (nitrogen source) were mixed to dope nitrogen into TiO2 and RGO simultaneously and form N-TG to remove HA from aqueous solution. To confirm the effect of the N-doping and determine the best N-doping ratio for N-TG, various characterization and HA removal tests using different samples were conducted, we found the best N-doping ratio is ~1.46 at.%. The influences of the initial HA concentration, temperature and pH on HA removal performance were measured and discussed, notably, temperature range of 25–35 °C and neutral solution are more fitable for HA removal. HA removal is in the synergistic effect of adsorption and degradation. The presence of RGO almost doubles the adsorption ability of the composite, which does have a significant improvement on HA removal efficiency. Pretreated in darkness has an extra 2% improvement on HA removal efficiency.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past 20 years, global warming, increased carbon emissions and acid subsidence have led to a significant increase of natural organic matter (NOM) in natural waters on a global scale [1] and caused a tremendous impact on water health. Because of the loose aggregation structure and strong adsorption complexation of NOM [2], the transition and transformation of particles and trace organic/inorganic pollutants in water are greatly influenced, the extent of the water pollution condition is difficult to estimate. NOM is the primary cause of water chromaticity and is readily transformed into various disinfection by-products which are mutagenic, teratogenic and carcinogenic [3]. NOM will increase the coagulant dosage during conventional water treatment, and then speed up pipeline corrosion in the water distribution process [4]. Thus, NOM removal is of great significance to improving water quality and ensuring the safety of drinking water.
The components of NOM are complex; the primary composition is humus (60–90%) which is usually divided into humic acid (HA), fulvic acid (FA) and humin (HU) [5]. Among these, HA is considered the typical material of NOM because HA has most of the same functional groups as NOM.
Strengthening the coagulation, adsorption, ion-exchange interaction, membrane separation and advanced oxidation [6,7,8,9,10] have been approaches used to reduce the HA concentration in water. Among all these processes, photocatalysis technology, which is a type of advanced oxidation technology, is efficient in reducing HA in water. Under the effects of light, a photocatalyst absorbs light energy and produces an electron–hole, and then hydroxyl free radicals (·OH) form on the surface of the photocatalyst. The ·OH in the presence of strong oxidizing agents can degrade HA to H2O, CO2 and other small molecular compounds [11, 12].
In various photocatalysts, TiO2 is the core of the photocatalysis technology because it is highly chemical resistant and non-toxic, and has a high catalytic activity, strong oxidation ability and low cost [13, 14]. However, TiO2 as a photocatalyst is not exactly perfect because certain disadvantages limit its further application. TiO2 can only be triggered by ultraviolet light (λ ≤ 387.5 nm) as the energy gap between the valence and conduction bands is ~3.2 eV, implying that only ~4% of sunlight has enough energy to trigger TiO2 [15, 16]. Additionally, the photogenerated electron–hole easily recombines if the electron is not transferred out in sufficient time [17]. A high electron–hole recombination rate means low quantum efficiency [18, 19]. Numerous studies have been devoted to reduce the width of the forbidden band and electron–hole recombination rate.
Graphene comprises a single sheet of sp 2-hybridized carbon atoms in a hexagonal lattice and has received increased attention in various industries because of its high surface area (2630 m2 g−1), and attractive electronic, mechanical, thermal and optical properties [20, 21]. As a derivative of graphene, graphene oxide (GO) is suitable for application in water treatment because of the hydrophilic oxygen-containing functional groups (e.g., carboxyl groups, hydroxyl groups, epoxy groups) [22]. GO is often used as an adsorbent, having a good effect on the removal of metals and organic matter [23, 24]. Notably, GO in high-temperature anoxic environment is often converted to reduced graphene oxide (RGO) which contains less oxygen-containing functional groups but owns higher charge carrier mobility than GO [25].When TiO2 combines with GO, an electron can be quickly conducted out by GO or RGO to avoid electron–hole combination. Additionally, Ti–O–C bonds will form and therefore expand the light absorption range [19, 26]. Studies show that the best amount of GO in a TiO2/RGO composite to promote photocatalytic performance is 5% [27].
Nitrogen-doping of TiO2, which replaces oxygen vacancies with nitrogen (Ns) or introduces nitrogen to TiO2 interstitially (Ni), is efficient in reducing the energy gap between the valence and conduction bands [28, 29]. Nitrogen-doping of TiO2 is considered a promising photocatalyst with increased photocatalytic efficiency. Nitrogen-doping makes sense not only for TiO2, but also for graphene. Studies show that nitrogen-doping of graphene can change the electronic structure of graphene [30], promote free carrier density [31], open the band gap and adjust the conductive type [32]. In photocatalysis, nitrogen-doping of graphene can improve the photocatalytic effect more than that of graphene [13, 33].
Predecessors have conducted significant research on TiO2, TiO2/graphene materials, nitrogen-doping of TiO2/graphene-family materials (both nitrogen-doping of TiO2 and nitrogen-doping of graphene-family materials) and their characteristics [13, 18, 19, 33]. Their work laid a solid foundation for further research. In this study, we adopted a hydrothermal method for mixing nitrogen sources, TiO2 and GO to dope nitrogen into TiO2 and RGO (N-TG) simultaneously, and the optimal nitrogen-doping amount and the form of nitrogen in N-TG were analyzed. Then, the HA removal ability of N-TG was tested; and the influence of the initial HA concentration, temperature and pH on the HA removal ability was investigated. Additionally, the synergistic effect of TiO2 and RGO on HA removal were discussed.
Materials and methods
Nanoparticles and chemicals
Anhydrous ethanol (purity ≥ 99.7%), tetrabutyl titanate (purity ≥ 98.5%), glacial acetic acid (purity ≥ 99.5%), hydrochloric acid (HCl, 6 M), urea (purity ≥ 99.0%), sodium hydroxide (purity ≥ 99.5%) were purchased from Sinopharm Chemical Reagent Co. Ltd., China. Humic acid (HA, purity ≥ 95.0%) was purchased from Alfa Aesar Co. Ltd., China. Graphene oxide (GO, purity ≥ 99.5%, powder, diameter = 0.5–5 μm, thickness = 0.6–1 nm) was purchased from Hengqiu Graphene Technology Co. Ltd., China. Deionized water (DI) used in this study was produced by a Milli-Q integral system.
Preparation of TiO2
TiO2 nanoparticles were prepared using a sol–gel approach. First, 10.0 mL tetra butyl titanate was slowly added to 40.0 mL anhydrous ethanol to form Solution A. Solution B was a mixture of 10.0 mL anhydrous ethanol, 4 mL DI and 2 mL glacial acetic acid. After the pH of Solution B had been adjusted to 2 using HCl, Solution B was added dropwise into Solution A with intense mixing. A further 0.5 h stirring was needed to ensure intensive mixing. Then, the mixture was settled for ~3 h to form a pale yellow transparent gel. A dry atmosphere at 100 °C was used to dry the mixture and form yellow crystals. The solids were transformed to a white powder by grinding. After calcining at 300 °C for 1 h and then at 450 °C for 2 h, the preparation of TiO2 was completed.
Preparation of nitrogen-doping TiO2/RGO (N-TG)
N-TG was prepared using a hydrothermal method. First, 0.05 g GO was dispersed in 70 mL DI with ultrasonic treatment for 2 h. A further 1-h ultrasonic treatment was needed after the addition of 0.95 g TiO2 and a certain amount of urea (the corresponding urea weights for N-TG is shown in Table 1). Then, the solution was moved to a 100 mL Teflon-lined stainless autoclave. N2 was transported to the solution to discharge other gases in the solution and keep a nitrogen balance in the reactor. The reactor was held in a high-temperature atmosphere at 200 °C for 12 h and cooled to room temperature naturally. Finally, the suspension was centrifuged at 5000 rpm followed by washing with DI at least three times, and then dried under vacuum at 80 °C. The TG was prepared by the same process but without urea.
Characterization of samples
Scanning electron microscopy (SEM, Hitachi SU8010, Japan) and transmission electron microscopy (TEM, JEM-2100F, Japan) equipped with element mapping equipment were used to characterize the morphology of the samples. X-ray diffraction (XRD, X'Pert PRO MPD, Netherlands) was used to characterize the crystal planes of TiO2 and other composites. The element compositions of the samples were analyzed by X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi, England). The diffuse reflectance absorption spectra (DRS) of the samples were in the range from 200 to 800 nm as detected by a UV–Vis spectrophotometer (Cary 5000, America). The photocurrents were measured using a standard three-electrode cell (RST5200, China). The zeta potential of the samples in solutions of different pH was measured by the Zetasizer Nano (Zetasizer Nano ZS90, England). The total organic carbon (TOC) of HA solutions is measured by the TOC analyzer (Aurora 1030W, America).
Preparation and measurement of HA solution
A 900-mL NaOH solution at pH = 11 was prepared first, and 1 g HA was dissolved in the solution with rapid mixing. Then, the pH of the solution was adjusted to 7 using HCl. DI was added to the solution to make the solution volume reach 1 L accurately. This HA solution was filtered through a 0.45-μm filter membrane to remove the insoluble substances. The insoluble substances (Mis, mg) were weighing after drying, and the concentration of the HA solution was calculated [c = (1000 − Mis) mg L−1]. The HA solution was kept out of the sun in the freezer at 4 °C before using.
In this study, we adopted UV254 to express the HA concentrations; thus, a concentration–absorbance standard curve was needed before the tests. The absorbance of the HA solutions which were diluted to 2.5, 7.5, 12.5, 25 and 50 mg L−1 was measured by ultraviolet and visible spectrophotometer at 254 nm. A fitting degree more than 0.999 was considered desirable in the concentration–absorbance standard curve. The concentration–absorbance standard curve had to be re-measured every time the solution impact factors (e.g., temperature and pH) were changed.
Results and discussion
Characterization and nitrogen-doping effect
Figure 1 shows the images of the SEM, TEM and element mapping for the as-prepared N-TG2 composite. RGO is on the sheet, while TiO2 is intensively distributed on both sides of the RGO as shown in Fig. 1a. Only a few parts of RGO were exposed, which is beneficial for utilizing a light source because exposed RGO will absorb light. However, the TiO2 particle is partly agglomerated, which may be due to the high-temperature atmosphere in the preparation, while the crystal planes of TiO2 are unchanged as shown in Fig. S1. The TEM image shown in Fig. 1b confirms the irregularity of TiO2 in size. The elemental mapping patterns of Ti (Fig. 1d), O (Fig. 1e), C (Fig. 1f) and N (Fig. 1g) corresponding to the boxed area in Fig. 1c are listed, the percentage of elements in composite are 56.97, 38.96, 3.29 and 0.78 wt%, respectively. The superior elements dispersion implies the success hybridization of TiO2 and RGO, and N-doping in the composite.

a SEM images of N-TG2, b, c TEM images of N-TG2; and elemental mapping patterns of d Ti, e O, f C and g N corresponding to the boxed area in (c)
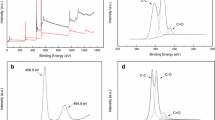
The prepared composites were investigated using XPS spectra to analyze the element compositions. Figure 2a shows the XPS spectra of N-TG2, the peaks at 38, 63, 285, 400, 459, 530 and 565 eV can be assigned to Ti 3p, Ti 3s, C 1s, N 1s, Ti 2p, O 1s and Ti 2s, respectively. The peak of C 1s as shown in Fig. 2b can primarily give credit to C=C bonds (284.8 eV), the peaks at 285.6, 286.6 and 289.6 eV could be assigned to C=N and C–O bonds, C=O and C–N bonds and other C–O bonds (such as Ti–O–C bonds). As doping may cause the change in electric charge distribution both in and out of the nucleus, the binding energy of elements may adjust slightly. Thus, the binding energy of elements and their bonds are not always the same but may be similar as has been previously reported [19, 33].

a XPS spectra of N-TG2, b C 1s XPS spectra of N-TG2; N 1s XPS spectra of c TG and d N-TG2
The N 1s XPS spectra in Fig. 2c show the slight peak at ~400 eV, which may be from N2 in the preparing of TG, but the amount was too small, while the ratio was almost zero in the elemental compositions of TG as shown in Table 1. The N content in N-TG1, N-TG2 and N-TG3 is also shown in Table 1. The ratio of N increased with the increase in urea. In the preparation of N-TG, though the doping amount of N was a few parts of the urea, most of the urea was transformed to NH3 and other nitrides in the high-temperature atmosphere during the preparation. The N 1s XPS spectra of N-TG2 as shown in Fig. 2d could be fitted to β-N (397.8 eV), γ-N and pyridinic N (399.3 eV), pyrrolic N (400.4 eV) and graphitic N (401.8 eV). The β-N could be assigned to Ns (Ti–O–N or/and Ti–N), while γ-N could be assigned to Ni (N2 or/and NO). The Ni is considered a promising doping form because it can form an impurity energy level which is ~0.75 eV above the valence band, while the impurity energy level made by Ns is ~0.14 eV above the valence band [28]. Obviously, the peak area of γ-N is larger than β-N, which is conducive to reducing the forbidden bandwidth.
Figure 3a shows the UV–Vis diffuse reflection spectra of TiO2, TG, N-TG1, N-TG2 and N-TG3. TiO2 had an absorption band lower than 387 nm, which is consistent with anatase TiO2. With the presence of RGO, a wider range of absorption band appears because of the broad absorption range of RGO [14, 34] and the Ti–O–C bonds that formed between TiO2 and RGO. The introduction of N into the TiO2/RGO composite has a further increase in the absorption range because of the formation of impurity energy levels in the TiO2 band structure. However, an increased N-doping amount does not increase absorptance results, as N-TG1 exhibits the widest absorption range among N-TG1, N-TG2 and N-TG3. We hypothesize that the best N-doping ratio to wider absorption range is in the range of 0–1.46 at.%. The corresponding band gap is shown in Fig. 3b, which is estimated from the plot of the transformed Kubelka–Munk function versus the light energy. The band gaps for TiO2, TG, N-TG1, N-TG2 and N-TG3 are 3.21, 3.11, 2.82, 2.86 and 2.96, respectively.

a UV–Vis diffuse reflection spectra of TiO2, TG, N-TG1, N-TG2 and N-TG3, b the corresponding plot of the transformed Kubelka–Munk function versus the light energy from (a)
Figure 4a shows the residual HA concentration when the initial concentration is 30 mg L−1 treated by TiO2, TG, N-TG1, N-TG2 and N-TG3 at 25 °C in UV light. The HA removal efficiency of TiO2 after being treated for 120 min was 38.3%. The HA could have been degraded completely given sufficient time, but the degradation rate was not satisfactory. With TiO2 and GO composite (TG), the HA removal efficiency was increased to 81.3%, verifying the immense effect of RGO on promoting the advantages of TiO2. While nitrogen was doped into the composite of TiO2 and RGO (N-TG), a further enhanced photocatalysis emerged. The HA removal efficiency was increased by at least 7% compared to that of TG, its efficiency is brilliant compared with other HA removal technologies reported previously [5, 7, 9]. This outcome is consistent with the result of the UV–Vis diffuse reflectance spectra as shown in Fig. 3 because the N-doping would reduce the forbidden bandwidth. Similar HA removal tests exposed in sunlight were tested, and the results are shown in Fig. 4b, all the efficiencies have improved compared with which exposed in UV light except TiO2, while the HA removal efficiency of N-TG1 owns the largest improvement which is almost the same with N-TG2 because N-TG1 owns the narrowest forbidden bandwidth.

HA removal performance of TiO2, TG, N-TG1, N-TG2 and N-TG3 in a UV light and b sunlight
Additionally, the N-doping gives the composite a higher electrical conduction ability as shown in Fig. 5. Though the forbidden bandwidth of N-TG2 was wider than N-TG1, its electrical conduction ability was faster, and thus the highest HA removal efficiency was made by the N-TG2, in which the N-doping ratio was 1.46 at.%. We hypothesize that the best N-doping ratio to increase the TG photocatalytic ability is ~1.46 at.%.

Transient photocurrent response for TiO2, TG, N-TG1, N-TG2 and N-TG3
Influences of initial HA concentration, temperature and pH
A series of tests with different initial HA concentrations treated by 1 g L−1 N-TG2 at 25 °C in UV light were conducted. The HA solutions were diluted to 10, 20, 30, 40 and 50 mg L−1 with the HA solution previously prepared without any other reagent, so the pH of the solutions was ~7. As shown in Fig. 6, the residual HA concentrations after being treated by N-TG2 for 120 min were 1.1, 1.9, 2.5, 3.1 and 3.7 mg L−1, and the corresponding removal efficiencies were 89.0, 90.5, 91.7, 92.3 and 92.6%. With the increase in the initial HA concentration, the residual HA concentration and HA removal efficiency increased simultaneously, which confirmed the pseudo-first-order kinetic model [16]. The pseudo-first-order kinetic models of the different initial HA concentrations were calculated and are listed in Table 2. When the initial HA concentration was 10 mg L−1, the coefficient of determination was more than 0.999, implying a good fitness of pseudo-first-order kinetic model. While the correlation coefficient does decrease with increasing initial HA concentration, the pseudo-first-order kinetic model maintains a high level of accuracy over the tested concentration range.

HA removal performance of N-TG2 in different initial HA concentrations
The influences of temperature were tested and are shown in Fig. 7a. In this test, 1 g L−1 N-TG2 was used to photodegrade the HA solution in which the initial concentration was 30 mg L−1 at 5, 15, 25 and 35 °C, the pH of the solution was adjusted to 7, and an ultraviolet lamp was used as the light source.

HA removal performance of N-TG2 at different temperatures in a UV light and b darkness
Because the activation energy for the photocatalytic reaction is not high, the influence of temperature on photocatalysis is small. Thus, the residual concentrations of the HA treated by N-TG2 after 120 min at different temperatures were not entirely different. However, temperature does affect the adsorption performance of N-TG, particularly RGO, which is always used as an adsorbent in water treatment. The residual HA concentrations after being treated by N-TG2 for 120 min at 5–35 °C indicated a downward trend. We hypothesize that the difference in concentration curves for the different temperatures was primarily caused by the adsorption performance of N-TG2. The difference was not noticeable because the amount of RGO was at such a low level.
To ensure the adsorption performance of N-TG2 across changing temperatures, a similar test was conducted in darkness. As shown in Fig. 7b, the HA removal efficiency increased with the increase in temperature from 5 to 25 °C, and the HA removal efficiency at 35 °C was similar to that at 25 °C. A photocatalyst cannot provide ·OH without a light source, and adsorption will become the primary cause for reducing the HA concentration in the solution. When temperatures increase, the molecular motion is more intense, leading to an increasing potential for effective contact, and therefore, a better adsorption effect. However, most of the adsorption belongs to the exothermic reaction [35], which means a high temperature is not beneficial for adsorption. Thus, an optimum temperature for N-TG2 to remove HA may exist, and we hypothesize that the optimum temperature is in the range of 25–35 °C.
The pH of natural water is generally in the range of 5–13 [36], so we adjusted the pH of the HA solution to 5, 7, 9, 11 and 13 to test the influence of pH on HA removal. In this test, 30 mg L−1 HA solutions with 1 g L−1 N-TG2 or TiO2, respectively, at 25 °C and various pH levels were exposed to UV light. We measured the residual HA concentrations that were being treated for 120 min and plotted a pH-removal efficiency changing curve as shown in Fig. 8a. After being treated by N-TG2 for 120 min, the HA removal efficiency in the solutions of pH = 5, 7, 9, 11 and 13 was 87.3, 90.7, 72.7, 58.0 and 48.7%, respectively. The HA removal efficiency of N-TG2 decreased with the increase in pH in neutral and alkaline solution, while the highest HA removal efficiency appears in the solutions of pH = 7. Several factors may be associated with this phenomenon. The rate of ·OH generation may be different under different pH. Besides, a photocatalyst has a particular pH called a point of zero charge (pHpzc) [37] in which the positive charge is equal to the negative charge in a solution. When the pH < pHpzc, the photocatalyst surface is in positive charge, when the pH > pHpzc, the photocatalyst surface is in negative charge. Additionally, the HA has many carboxyl and hydroxyl groups, lending electronegativity to the solution at 5 < pH < 13, and its pHpzc is 1.8 as shown in Fig. 8b. Furthermore, the solubility of HA in solutions of various pH levels is different [38], and HA does not dissolve in a solution of lower pH so it may be easier to remove.

a HA removal performance of TiO2 and N-TG2 at various pH levels, b the zeta potential of 1 g/L TiO2, N-TG2 and HA at various pH levels
The process of ·OH generation for N-TG may be summed up into the above Eqs. (1)–(6). Alkaline pH is more suitable for holes to oxidize OH− or H2O and form free ·OH, while acidic pH will promote electrons react with dissolved oxygen to produce ·O2 − and ·OH successively. Obviously, neutral and alkaline solution is beneficial for ·OH generation.
The pHpzc of N-TG2 and TiO2 were tested and shown in Fig. 8b. The pHpzc of N-TG2 was 4.3, which was decreased by 2.1 compared with TiO2, and oxygen-containing functional groups in RGO were responsible for the decrease. In the solution at pH range 5–13, the N-TG2 surface is in negative charge and HA is also in negative charge. Thus, they cannot easily be in proximity to each other because of electrostatic repulsion. The electrostatic repulsion increased with the increase in pH because the negative charge of both N-TG2 and HA was increased. Meanwhile, the HA adsorbed by N-TG2 stopped HA in solution from closing to the N-TG2 because of the electrostatic repulsion, which will increase with the pH likewise.
TOC test
UV254 represents the chromogenic substances in HA and is not typical enough to show the mineralization of the pollutants. TOC is a comprehensive indicator of the total amount of organic matter in water. The TOC of the solution was measured, which the initial HA concentration is 30 mg L−1. The result is shown in Fig. 9. The removal efficiency of TOC is always slightly lower than HA concentration, implying that most of HA was mineralized into inorganic small molecular compounds, while part of HA changed to other organic carbon. Similar result has been reported by Yuan [39], who prepared Fe–Zn co-doped TiO2 and found the TOC removal efficiency was lower than that of UV254 because of the presence of intermediates.

HA concentration and TOC removal efficiency curves treated by N-TG2 in UV light
Synergistic effect of TiO2 and RGO
In the process of testing the influence of temperature on HA removal, we find that adsorption performance is one of the influential factors in HA removal efficiency. With the effect of adsorption, the HA photodegradation rate is faster. It is assumed that the process of photocatalysis contains mass transfer, adsorption, degradation and desorption [40, 41]. For the N-TG composites, degradation primarily relies on TiO2, while RGO greatly improves the adsorption, i.e., HA removal is in the synergistic effect of TiO2 and RGO.
An experiment to test the adsorption capacity of TiO2 and N-TG2 was conducted. In this test, 1 g L−1 N-TG2 or TiO2 was distributed in 30 mg L−1 HA solution of pH = 7 at 25 °C in darkness, and N-TG2 in UV light was performed under the same conditions. The adsorption efficiency is shown in Fig. 10. The TiO2 absorbed 2.8 mg L−1 HA after 120 min, while N-TG2 absorbed 5.4 mg L−1. Because the mass fraction of RGO in N-TG2 is only ~5%, the adsorption capacity increased by RGO is ~52 mg g−1 in theory. However, adsorption was not sufficient for N-TG to remove HA because its adsorption ability was limited due to an inherent adsorption equilibrium, which means it cannot adsorb HA continuously. As shown in Fig. 10, the removal efficiency of N-TG2 is 18.0%, which would increase to 91.7% with the synergistic effect of degradation.

HA adsorption performance of TiO2 and N-TG2 in darkness, and HA removal performance of N-TG2 in UV light
To ensure the performance of adsorption in HA removal, two groups of 1 g L−1 N-TG2 were distributed in 30 mg L−1 HA solution of pH = 7 at 25 °C in UV light with one group first being treated in darkness for 60 min, and the result is shown in Fig. 11. With the darkness treatment, the HA removal efficiency was increased compare to that without pretreatment, indicating the important role of adsorption in HA removal. However, the pretreatment in darkness helped to slightly increase the efficiency, it would be better for an extra hour treatment under UV light. Besides, too much HA absorbed on the surface of N-TG composite may hinder the degradation because the N-TG cannot receive enough light energy, which may explain the gradual lower trend that appeared in the first 5 min as shown in Fig. 11.

HA removal performance of N-TG2 with and without pretreatment in darkness
A model of N-TG is shown in Fig. 12. In N-TG composite, TiO2 is the basic material, and the adding of RGO reduces the forbidden bandwidth and transfers electrons rapidly. In the photocatalysis process, RGO greatly strengthens the composite for HA adsorption and promotes degradation efficiency naturally; TiO2 degrades HA, which is adsorbed on the surface of the composite. Thus, adsorption would never reach equilibrium, and the composite would absorb HA continuously. The HA is removed more efficiently with the synergistic effect of TiO2 and RGO.

Schematic diagram of N-TG
Conclusions
In this study, GO, TiO2 and urea (nitrogen source) were mixed to dope nitrogen in TiO2 and RGO simultaneously and formed N-TG. Using XPS, UV–Vis, transient photocurrent response and HA removal tests, it was found that N-doping reduces the forbidden bandwidth and transfers electronic rapidly, with 1.46 at.% being the best N-doping ratio for N-TG which the band gap is 2.86. When studying the effect of the initial HA concentration, it was confirmed that the HA removal process conforms to the pseudo-first-order kinetic model, while lower initial HA concentrations have a slight better fit. The influence of temperature on HA removal was understood by observing the adsorption ability in different temperatures, and the most appropriate temperature may be in the range of 25–35 °C. A higher pH meant faster ·OH generation rate but stronger electrostatic repulsion; thus, the HA removal was the best at pH = 7. TOC test confirms the mineralization of most of HA, while intermediates exist meanwhile. Further studies to test the effect of adsorption were conducted to confirm the synergistic effect of TiO2 and RGO on HA removal. The removal efficiency of N-TG2 in darkness is 18.0%, which would increase to 91.7% with the synergistic effect of photodegradation; pretreated in darkness has an extra improvement on HA removal efficiency. In general, N-TG was efficient in removing the HA from aqueous solution and is promising for further application in water treatment.
References
Delpla I, Jung AV, Baures E, Clement M, Thomas O (2009) Impacts of climate change on surface water quality in relation to drinking water production. Environ Int 35:1225–1233
Wu X, Tan X, Yang S, Wen T, Guo H, Wang X, Xu A (2013) Coexistence of adsorption and coagulation processes of both arsenate and NOM from contaminated groundwater by nanocrystallined Mg/Al layered double hydroxides. Water Res 47:4159–4168
Jiang Y, Goodwill JE, Tobiason JE, Reckhow DA (2016) Impacts of ferrate oxidation on natural organic matter and disinfection byproduct precursors. Water Res 96:114–125
Broo AE, Berghult B, Hedberg T (1998) Copper corrosion in water distribution systems—the influence of natural organic matter (NOM) on the solubility of copper corrosion products. Corros Sci 40:1479–1489
Zhang J, Gong J, Zenga G, Ou X, Jiang Y, Chang Y, Guo M, Zhang C et al (2016) Simultaneous removal of humic acid/fulvic acid and lead from landfill leachate using magnetic graphene oxide. Appl Surf Sci 370:335–350
Xu H, Jiao R, Xiao F, Wang D (2014) Effects of different coagulants in treatment of TiO2-humic acid (HA) water and the aggregate characterization in different coagulation conditions. Colloid Surf A 446:213–223
Dong C, Chen W, Liu C (2014) Preparation of novel magnetic chitosan nanoparticle and its application for removal of humic acid from aqueous solution. Appl Surf Sci 292:1067–1076
Lin T, Lu Z, Chen W (2015) Interaction mechanisms of humic acid combined with calcium ions on membrane fouling at different conditions in an ultrafiltration system. Desalination 357:26–35
Thuyavan YL, Anantharaman N, Arthanareeswaran G, Ismail AF (2014) Adsorptive removal of humic acid by zirconia embedded in a poly(ether sulfone) membrane. Ind Eng Chem Res 53:11355–11364
Lim SM, Chiang K, Amal R, Fabris R, Chow C, Drikas M (2007) A study on the removal of humic acid using advanced oxidation processes. Sep Sci Technol 42:1391–1404
Lee PF, Sun DD, Leckie JO (2007) Adsorption and photodegradation of humic acids by nano-structured TiO2 for water treatment. J Adv Oxid Technol 10:72–78
Birben NC, Uyguner-Demirel CS, Sen Kavurmaci S, Gurkan YY, Turkten N, Cinar Z, Bekbolet M (2017) Application of Fe-doped TiO2 specimens for the solar photocatalytic degradation of humic acid. Catal Today 281:78–84
Liu Y, Pei F, Lu R, Xu S, Cao S (2014) TiO2/N-graphene nanocomposite via a facile in situ hydrothermal sol–gel strategy for visible light photodegradation of eosin Y. Mater Res Bull 60:188–194
Zhang Y, Pan C (2011) TiO2/graphene composite from thermal reaction of graphene oxide and its photocatalytic activity in visible light. J Mater Sci 46:2622–2626. doi:10.1007/s10853-010-5116-x
Chen X, Mao SS (2007) Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications. Chem Rev 107:2891–2959
Chen W, Ye T, Xu H, Chen T, Geng N, Gao X (2017) An ultrafiltration membrane with enhanced photocatalytic performance from grafted N-TiO2/graphene oxide. Rsc Adv 7:9880–9887
Johra FT, Jung W (2015) RGO–TiO2–ZnO composites: synthesis, characterization, and application to photocatalysis. Appl Catal A Gen 491:52–57
Pelaez M, Nolan NT, Pillai SC, Seery MK, Falaras P, Kontos AG, Dunlop PSM, Hamilton JWJ et al (2012) A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl Catal B Environ 125:331–349
Wang R, Wu Q, Lu Y, Liu H, Xia Y, Liu J, Yang D, Huo Z et al (2014) Preparation of nitrogen-doped TiO2/graphene nanohybrids and application as counter electrode for dye-sensitized solar cells. Acs Appl Mater Inter 6:2118–2124
Li Z, Zhang H, Liu Q, Sun L, Stanciu L, Xie J (2013) Fabrication of high-surface-area graphene/polyaniline nanocomposites and their application in supercapacitors. Acs Appl Mater Inter 5:2685–2691
Meyer JC, Geim AK, Katsnelson MI, Novoselov KS, Booth TJ, Roth S (2007) The structure of suspended graphene sheets. Nature 446:60–63
Thuy-Duong N, Viet HP, Shin EW, Hai-Dinh P, Kim S, Chung JS, Kim EJ, Hur SH (2011) The role of graphene oxide content on the adsorption-enhanced photocatalysis of titanium dioxide/graphene oxide composites. Chem Eng J 170:226–232
Duru I, Ege D, Kamali AR (2016) Graphene oxides for removal of heavy and precious metals from wastewater. J Mater Sci 51:6097–6116. doi:10.1007/s10853-016-9913-8
Xue Z, Zhao S, Zhao Z, Li P, Gao J (2016) Thermodynamics of dye adsorption on electrochemically exfoliated graphene. J Mater Sci 51:4928–4941. doi:10.1007/s10853-016-9798-6
Jia Z, Li H, Zhao Y, Frazer L, Qian B, Borguet E, Ren F, Dikin DA (2017) Electrical and mechanical properties of poly(dopamine)-modified copper/reduced graphene oxide composites. J Mater Sci 52:11620–11629. doi:10.1007/s10853-017-1307-z
Liu S, Sun H, Liu S, Wang S (2013) Graphene facilitated visible light photodegradation of methylene blue over titanium dioxide photocatalysts. Chem Eng J 214:298–303
Pan X, Yang M, Tang Z, Xu Y (2014) Noncovalently functionalized graphene-directed synthesis of ultralarge graphene-based TiO2 nanosheet composites: tunable morphology and photocatalytic applications. J Phys Chem C 118:27325–27335
Qian W, Greaney PA, Fowler S, Chiu S, Goforth AM, Jiao J (2014) Low-temperature nitrogen doping in ammonia solution for production of N-doped TiO2-hybridized graphene as a highly efficient photocatalyst for water treatment. Acs Sustain Chem Eng 2:1802–1810
Li J, Chen LX, Li X, Zhang CC, Zeng FL (2015) Hollow organosilica nanospheres prepared through surface hydrophobic layer protected selective etching. Appl Surf Sci 340:126–131
Barrejon M, Primo A, Gomez-Escalonilla MJ, Fierro JLG, Garcia H, Langa F (2015) Covalent functionalization of N-doped graphene by N-alkylation. Chem Commun 51:16916–16919
Lherbier A, Botello-Mendez AR, Charlier J (2013) Electronic and transport properties of unbalanced sublattice N-doping in graphene. Nano Lett 13:1446–1450
Meng F, Li J, Cushing SK, Zhi M, Wu N (2013) Solar hydrogen generation by nanoscale p–n junction of p-type molybdenum disulfide/n-type nitrogen-doped reduced graphene oxide. J Am Chem Soc 135:10286–10289
Liu C, Zhang L, Liu R, Gao Z, Yang X, Tu Z, Yang F, Ye Z et al (2016) Hydrothermal synthesis of N-doped TiO2 nanowires and N-doped graphene heterostructures with enhanced photocatalytic properties. J Alloy Compd 656:24–32
Dawlaty JM, Shivaraman S, Strait J, George P, Chandrashekhar M, Rana F, Spencer MG, Veksler D et al (2008) Measurement of the optical absorption spectra of epitaxial graphene from terahertz to visible. Appl Phys Lett 93:193–197
Zhang H, Gu W, Li M, Fang W, Li Z, Tao W (2015) Influence of environmental factors on the adsorption capacity and thermal conductivity of silica nano-porous materials. J Nanosci Nanotechnol 15:3048–3054
Pierrard JC, Rimbault J, Aplincourt M (2002) Experimental study and modelling of lead solubility as a function of pH in mixtures of ground waters and cement waters. Water Res 36:879–890
Kosmulski M (2002) The significance of the difference in the point of zero charge between rutile and anatase. Adv Colloid Interface 99:255–264
Kipton H, Powell J, Town RM (1992) Solubility and fractionation of humic acid: effect of pH and ionic medium. Anal Chim Acta 267:47–54
Yuan R, Zhou B (2016) Effect of ion (Al, Fe and Zn) co-doped TiO2 nanotubes on photocatalytic degradation of humic acids under UV/ozonation for drinking water purification. Water Sci Tech W Sup 16:237–244
Hidaka H, Honjo H, Horikoshi S, Serpone N (2003) Photocatalyzed degradations on a TiO2-coated quartz crystal microbalance. I. Adsorption/desorption processes in the degradation of phenol and catechol. New J Chem 27:1371–1376
Zhou Q, Zhong Y, Chen X, Liu J, Huang X, Wu Y (2014) Adsorption and photocatalysis removal of fulvic acid by TiO2-graphene composites. J Mater Sci 49:1066–1075. doi:10.1007/s10853-013-7784-9
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 51678213 and 51578209), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ye, T., Chen, W., Xu, H. et al. Preparation of TiO2/graphene composite with appropriate N-doping ratio for humic acid removal. J Mater Sci 53, 613–625 (2018). https://doi.org/10.1007/s10853-017-1509-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1509-4