
Abstract
In this review, the regularities of formation, structure and high reactivity of two types of energetic metal/solid oxidizer nanocomposites (Al(Mg)/X (X = MoO3, (–C2F4–) n )) prepared by mechanoactivation are examined. One reason for the high reactivity is an increase in contact surface between the components occurring after mechanoactivation. Two methods for determination of area of contact surface S C between the components are used, and the values of S C for all the systems are estimated. Considerable attention is paid to the role of highly reactive defects (grain sizes, dislocations and stacking faults, paramagnetic centers, “weakly bound” oxygen in MoO3, etc.), formed in the components under mechanical stress. For the Me/MeO3 systems, the formation of point defects in the oxide is an important factor. It was found that, after mechanoactivation, the evolution of O2 from MoO3 occurs at 230–450 °C. It is argued that this process is associated with the thermal destruction of “weak” Mo–O bonds in the “bridge” oxygen. It was suggested that the formation of defect structure in MoO3 and increasing of the oxygen mobility under heating give rise to a low-temperature peak in DSC curves and initiated self-ignition on the fuel–air mixture. For composites Mg/MoO3, self-ignition occurs at temperature 100 °C lower than that for Al/MoO3: The decreasing of temperature can be connected with larger S C in the first system. In the Mg/(–C2F4–) n system, the reactions of magnesium defects with (–C2F4–) n are accompanied by a weak heat evolution, too low to initiate ignition. In this case, the reaction is initiated by the thermal depolymerization of (–C2F4–) n , while a high values of S C provide a complete conversion. In the case of shock-wave initiation, defects in the components play only a minor role in the conversion, whereas the value of S C remains to be highly important.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A standard way of improving the properties of energetic compositions, such as thermites, is to reduce the particle size of the components and increase the surface area of contact between the reactants. The contact surface area can be increased by a variety of methods, such as mixing the starting fine particles (including nanosized) mechanically and by sonication, sol/gel techniques, vacuum deposition and use of nanofoils (see, e.g., Ref [1]). Note that the so-called metastable intermolecular nanocomposites [2] are brought to the stage of practical applications [3].
One of the alternative methods for producing reactive metal/solid oxidant composites with a large contact surface area is their mechanoactivation (MA) in energy-intensive activators [4,5,6]. A key distinction of mechanochemical methods is that the formation of nanosized components involves the destruction of the material and rupture of chemical bonds at room temperature. These processes produce nanoparticles and nanocomposites with metastable defects of different natures (see, e.g., Ref. [7,8,9,10]).
The possibility of preparing nanosized aluminum by MA in the presence of carbon was demonstrated in the early 2000s [4, 11]. It turned out that nanoaluminum prepared by this method is highly reactive with respect to oxygen [12], water [13], carbon [14] and other substances [15]. Almost at the same time, attempts were made to create the mechanically activated energetic composites (MAEC) comprised of a metal and a solid oxidant [5, 6, 16,17,18]. In the last fifteen years, this direction has been developing in the USA, Russia and other countries, resulting in hundreds of original papers (see, e.g., Ref [19,20,21,22,23,24,25]) and several reviews and a book [1, 15, 26,27,28,29,30]. In particular, it turned out to be possible to increase the burning rate by several orders of magnitude and to realize steady detonation-like process at velocities of up to 1300 m/s [18, 27]. A differential scanning calorimetry (DSC) analysis made it possible to gain insights into the kinetics of the oxidation of aluminum with oxygen from the gas phase and from solid oxidizers [23, 28]. Based on this kinetic analysis, attempts were undertaken to predict the characteristics of ignition in different experimental conditions. However, this analysis ignores the possible existence of highly reactive defects in components formed by MA.
In the present paper, we analyze the defective structure of four Me/X MAEC (Me = Al, Mg; X = MoO3, (–C2F4–) n ). We estimate the value of the contact surface area between the components, types, and concentration of different defects in activated components. We tried to deduce the role of defects and the contact surface area on the promotion of some processes (self-ignition of air–fuel suspensions, heating in DSC measurements, detonation-like mode). Most of the primary experimental results have recently been published [31,32,33,34,35,36,37]. However, a joint analysis of the results is performed for the first time here. In these articles, the “degree” of mechanical activation is characterized by specific dose D (kJ/g) of applied mechanical energy. Dose was calculated by equation D = J·t, where J (W/g) is the specific power of mill in the conditions of activation and t is the duration of activation. The methods of J measurements are described in [38].
Preparation procedures, contact surface and defective structure in the Al(Mg)/(–C2F4–) n and Al(Mg)/MoO3 MAEC
Mechanoactivation of a mixture of two substances involves destruction and mixing of the components, as well as aggregation processes, accompanied by the formation of composites. This treatment changes the particle size of the components and increases the contact surface between the aggregate components and the concentration of various defects in the components. Optimal conditions for MA make it possible to create a composite with the highest reactivity, in which, however, chemical transformations during MAEC preparation have not yet occurred. The optimal conditions for MA are selected empirically by measuring the parameters of the subsequent chemical conversion, initiated by heat, shock wave or other impacts, which manifest themselves through the maximum values of the detonation or burning velocity, brightness temperature of combustion, etc. The data given below on the contact surface in the composite and on the defective structure correspond to MAEC prepared using an optimal activation dose.
System Al/(–C2F4–) n and Mg/(–C2F4–) n
After mechanoactivation of systems Al/(–C2F4–) n , their diffractograms [32] have broadened diffraction lines belonging to the two components, but no lines of other phases appeared. The broadening of the diffraction lines of Al was analyzed by the Williams–Hall method. Details are discussed in Ref [32]. It was found that broadening is caused by deformation, and value of microstrain was ε = 0.16%. Therefore, the grain sizes (regions of coherent scattering) were L ≥ 150 nm. From the value of ε, the dislocation density ρ d was estimated [32]. All these parameters are presented in Table 1. From the broadening of the lines of the second component (–C2F4–) n , one can suggest that the MA produces a fine polymer fraction with a grain size of about a few nanometers. It is known that the mechanical activation of (–C2F4–) n is accompanied by the rupture of polymer chains and the formation of free radicals of different structures. However, free radicals are thermally unstable and dissociate when heated to room temperature [39].
On the scanning electron microscopy (SEM) image of MAEC Al/(–C2F4–) n (Fig. 1a), one can see the particles with micron sizes L 1 and elongated polymer cords. From the more detailed analysis of structure of composites by the method of atom force microscopy (AFM) (Fig. 1b), it is clear that micron size (L 1) particles are the aggregates of the smaller particles with L 2 ~ 100–150 nm.

a SEM and b AFM images of mechanochemically activated composites Al/(–C2F4–) n
In Fig. 2, one can see the kinetics of the hydrogen release upon the interaction of Al with alkali. For the initial sample (1), the reaction was completed after 5–10 min and the activity (relative amount of interacted Al) was 96%. For MAEC (2), the reaction ended after about 1500 min (25 h) and the maximum conversion is equal to 32%. Incomplete dissolution of Al for MAEC was confirmed by the diffraction analysis [32]. It is natural to suggest that the difficulty in reaction occurrence and low conversion is explained by the encapsulation of Al particles by polytetrafluoroethylene (PTFE) layer, which hinders the interaction between metal and alkali.

Kinetics of dissolution of (1) initial aluminum and (2) aluminum in mechanically activated mixtures of Al/(–C2F4–) n
The existence of a core/shell structure is also confirmed by a number of other observations. Energy-dispersive spectroscopy (EDS) measurements are indicative of the homogeneity of the distributions of metal and fluorine atoms [32]. Atomic force microscopy (AFM) images taken in the topography and phase-contrast modes are identical [32], an observation indicative of a single-phase surface, taking into account significantly different coefficients of friction of aluminum and PTFE.
Thus, particles of MAEC Al/(–C2F4–) n have typical size L 1 close to ~5 micron. They are aggregates of finer grains with typical size L 2 ~ 100–150 nm. Moreover, aluminum particles are encapsulated by (–C2F4–) n shells. It is not clear whether there is a layer of PTFE between small particles of Al with L 2 = 100–150 nm.
Since the aluminum particles are encapsulated in (–C2F4–) n shells, the specific surface area of the contact between the components S C can be estimated. Sc is larger than the specific surface of aluminum particles with diameter L 1 and smaller than the specific surface area of aluminum grain with diameter L 2. Therefore, the value of S C can be estimated as 6/(ρL 1)K ≤ S C ≤ 6/(ρL 2)K, where ρ is the density, L 1 and L 2 are the particle and grain sizes for the aluminum component, and K is the weight fraction of aluminum in the mixture. At L 1 = 5 micron, S (min)C is equal to 0.12 m2/g; at L 2 = 100 nm, S (max)C is equal to 6 m2/g. In all cases, S C is smaller than 6 m2/g.
Similar data for the Mg/(–C2F4–) n system are listed in Table 1. The contact surface area and the size of the CSR for the magnesium component were approximately the same as those for the aluminum component. However, in this case, a detailed analysis of the anisotropy of the broadening of the X-ray lines of the magnesium component [33, 34], along with randomly distributed dislocations, shows the formation of basal and prismatic stalking faults in the magnesium (see Ref. [33] for details). The respective concentrations are given in Table 1. Thus, MA of the composite is accompanied by the decrease in the magnesium particle size by two orders of magnitude, encapsulation of submicron magnesium into fluoroplastic shells (with a specific surface area of contact S C up to 6 m2/g), and accumulation in the magnesium of randomly distributed dislocations (concentration up to 6 × 1010 cm−2), basal and prismatic stalking faults (with the highest probabilities of formation of 2.1 and 1.4%, respectively), and boundaries of coherent scattering regions. The PTFE undergoes the disordering and partial amorphization of the structure.
Systems Al/MoO3 and Mg/MoO3: The defective structure of MoO3 formed during mechanoactivation [36, 37]
Usually, the structure of orthorhombic α-MoO3 is described using irregular oxygen octahedral surrounding molybdenum atoms. According to alternative “striplike polymer model” [40], “polymer” MoO2–O–MoO2–O– chains are the main structural elements of a crystal (Fig. 3). Let us denote the non-equivalent oxygen atoms in MoO2 as terminal atoms O(1) and O(2) and bridge atoms as O(3). The lengths of the corresponding bonds [40] are d(Mo=O(1)) = 1.67 Å, d(Mo=O(2)) = 1.73 Å, d(Mo–O(3)) = 1.95 Å. Bridge oxygen atom O(3) is the “weakest” [40].

The “striplike polymer model” of MoO3 structure
Interacting via Mo and bridge oxygen O(3), two polymer chains form a “striplike” structure. Being located over one another, “strips” form bilayer “plate.” In this situation, the “strips” are bonded with one another via Mo and O(2), while oxygen O(1) is located on the “plate” surface. The plates interact with each other by means of the Van der Waals forces. A unit of cell accommodates two plates; lattice parameter a = 13.89 Å.
It was found [36] that the mechanoactivation of MoO3 proceeds in two stages. In the initial stage of the process, MoO3 particles are broken, so the MoO3 particle size decreases to tens of nanometers (curve 1 in Fig. 4), the specific surface area increases linearly up to 17–30 m2/g, the grain sizes reduce to 20–30 nm, and microstrain ε increases up to 0.6% [36]. In the second stage, at doses above 1 kJ/g, the basic process is the friction of the particles and their aggregation, accompanied by a decrease in the specific surface area, as well as the phase transition of MoO3 from the orthorhombic to the monoclinic modification. On the friction stage, grain sizes do no change, but microstrain increases up to 1%.

Decrease in the mean size of MoO3 <D> after its (1) individual MA and (2) MA in mixtures with Al
Figure 5 illustrates fragments of Raman spectra for the initial and mechanically activated MoO3 at the stages of fracture and friction. According to Ref [41], the spectral bands 1002–1007 cm−1 (1), 831–833 cm−1 (2) and 667–673 cm−1 (3) are assigned to three types of oxygen atoms Mo=O(1), Mo=O(2) and O(3)–Mo–O(3), respectively.

Fragments of Raman spectra of initial and activated MoO3. D is dose of MA. Frequencies corresponding to vibrations of group with terminal O(1), O(2) and bridge O(3) oxygen atoms are marked as 1, 2 and 3, respectively
The dependencies of the width of these three lines on the duration of MA are presented in Fig. 6. As the MA dose is increased, the Raman bands widen and shift toward longer wavelengths. For terminal atoms Mo=O(1), located on the “plate” surface, these changes are minimal. For terminal atoms Mo=O(2), the band width increases twice on the stage of fracture (up to 1 kJ/g) and then decreases on the stage of friction. Finally, for O(3)–Mo–O(3) bridge bonds the band width increases at both stages of fracture and friction while its shift is largest and amounts to 0.7%.

Half-widths of Raman spectral bands for vibration of groups with terminal О(1), О(2) and bridge О(3) oxygen atoms as a function of dose D of MoO3 activation
It is known [42] that widening and shift of the bands seem to be due to (1) a reduction in the grain sizes (surface factor) or (2) the appearance of local strains, point defects, etc. Surface factor can play a significant role, as changes in the width and the positions of the bands occur at the stage of fracture of MoO3 particles. Nevertheless, the band widening for O(3)–Mo–O(3) bridge bonds is observed on the stage of friction too, when surface area decreases. So we propose that additional band widening can be associated with deformation of “polymer” chain O(3)–Mo–O(3). This speculation is in agreement with the increase in microstrain ε.
Electron paramagnetic resonance (EPR) spectra for mechanically activated MoO3 (Fig. 7) combine three signals from different paramagnetic centers (PMC). The main contribution to the spectra of MA MoO3 is made by signal A with parameters: g 1 = 1.96, g 2 = 1.94, g 3 = 1.87. According to Ref. [43,44,45], this signal is due to ion Mo5+. Such PMC may be formed as a result of the rupture of Mo–O bridge bond in the chain O(3)–Mo–O(3).

EPR spectra for МА МоО3. Dose D = 0.8 kJ/g. Temperature is 77 K
The second signal is signal C (Fig. 7), which has narrow doublet with g = 1.975. This signal attributed to ion Mo3+ [45], or to proton disposed near Mo5+ [43]. The narrow isotropic signal E with g = 2.003, according to Ref. [43], points to the presence of free electrons in near surface layer of MoO3. Signals C and E have insignificant intensity compared to signal A.
The concentration of PMC increases during both fracture and friction stages. The maximum concentration is equal to 1 × 1018 spin/g. A linear correlation was found between concentration of PMC and microstrain ε [36].
Thus, the formation of MoO3 nanoparticles at the stage of fraction and their activation in the friction mode are accompanied by the formation of weakly bound oxygen (detected by Raman scattering spectroscopy) and broken molybdenum–oxygen bonds (paramagnetic centers Mo5+). When activated MoO3 is heated from 230 to 450 °C, defects disappear and molecular oxygen is released (Fig. 8). The release of oxygen was confirmed by a direct method and by detecting the presence of the Mo4O11 phase (4MoO3 → Mo4O11+O) after oxygen release. Details are available in Ref. [37].

Molecular oxygen evolution curve for MA MoO3. The arrows indicate the threshold temperatures of autoignition of (1) Mg/MoO3 and (2) Al/MoO3 (see “Promotion of self-ignition of the fuel−air mixtures by MAEC” section)
The mechanical activation of the Al/MoO3 and Mg/MoO3 systems was carried out in two steps [35]. In the first step, under MA of the initial oxide in an inert medium, nanosized n-MoO3 with a specific surface area of ~17 m2/g was prepared. In the second step, MA of Al(Mg) + n-MoO3 in a hexane media was performed. The liquid media were used to prevent an explosive reaction between the components during MA. The choice of this method of activation was motivated by the fact that during MA of a mixture of coarse MoO3 with of Al, the destruction of MoO3 particles proceeds markedly slower than that of the pure oxide (compare curves 1 and 2 in Fig. 4). The burning velocities for the co-activated systems turned out to be much lower than that for the two-step preparation, which confirms the validity of the method.
Figure 9 shows how the specific surface area of two-component mixtures changes in the course of their MA. Before activation, the specific surface of the mixture is almost equal to the partial contribution from the surface of the n-MoO3 component, since the contribution from the coarse metal powder to the specific surface area is small. As can be seen, in the course of MA, the specific surface of the mixture decreases. This suggests that the MA is accompanied by the aggregation of the mixture components. SEM examination of the Mg/MoO3 systems (Fig. 10a) confirmed that the size of the magnesium particles decreases from tens of microns to hundreds of nanometers and that submicron aggregates of Mg–MoO3 are formed with the component ratio close to that of starting mixture aggregates [35]. Thus, the decrease in the specific surface area ΔS upon MA can be interpreted as the upper estimate of the surface area of contact between the components (Sc ≤ ΔS). The corresponding value, Sc ≤ 8.5 m2/g, is given in Table 1.

Specific surface area S of (1) Mg/MoO3 and (2) Al/MoO3 mixtures as a function of the MA dose D

SEM images of mechanochemically activated composites a Mg/MoO3 and b Al/MoO3
For the Al/MoO3 system, the reduction in the specific surface of the composite upon MA is much smaller (Line 2 in Fig. 9). This correlates with the SEM data (Fig. 10b), which shows that a significant contribution comes from micron-sized Al flake particles coated with MoO3 nanoparticles.
After MA of the Mg/MoO3 mixture, the grain sizes in the magnesium are larger than 150 nm [34], while the formation of packing defects in the magnesium could not be detected because of the overlapping of the diffraction lines of Mg and MoO3. The aluminum in the activated MoO3 mixtures is characterized by a grain size of ~5–10 nm and by a low concentration of dislocations (Table 1).
Promotion of self-ignition of the fuel–air mixtures by MAEC
The idea that motivated this part of the work was to verify whether mechanically activated energetic composites (MAEC) can accelerate the self-ignition of fuel suspensions in air. These studies were carried out jointly with Professor K.Ya. Troshin from Institute of Chemical Physics of Russian Academy of Science.
MAEC Mg/n-MoO3, Al/n-MoO3, and Mg/PTFE with concentration of components as in Table 1 were prepared as in “Preparation procedures, contact surface and defective structure in the Al(Mg)/ (−C2F4−)n and Al(Mg)/MoO3 MAEC” section. The liquid hydrocarbon fuels were diesel oil and n-heptane into which MAEC powders were introduced to form suspensions. The concentration of additives in fuel is equal to 2.5 wt%. The ignition delay time of the fuel–air suspensions was measured at temperatures from 560 to 660 K and pressures of 1.4–1.5 atm.
The experiments were performed in a rapid mixture injection static reactor (Fig. 11) with 12-cm spherical-cylindrical stainless-steel chamber, which can be disassembled. During the experiment, the reactor was evacuated, filled to atmospheric pressure with air and heated up to a certain temperature.

Schematic diagram of the rapid mixture injection setup: (9) high-pressure chamber, (10) electromagnetic valve, (11) U-shaped tube with the sample. Reproduced from [31] with permission from Pleiades Publishing, Ltd
The test slurry fuel (without or with additives of MAEC) was placed in U-shaped tube (11 in Fig. 11); one end of which was open to the reactor, whereas the other end was connected through an electromagnetic valve (10 in Fig. 11) with high-pressure chamber 9. The amount of fuel supplied through the U-shaped tube was such as to produce a specific stoichiometric mixture with air in the reactor. Once the valve is open (1 in Fig. 12), fuel dispersed in the cold air stream was brought into reactor heated to a predetermined temperature and mixed with the air present therein. The valve remained open during 0.2 s. Previous temperature measurements showed that this time is sufficient to heat room-temperature air injected into reactor to the reactor wall temperature and to form a homogeneous mixture. The ignition delay time of fuel–air mixture was measured as time interval from the end of fuel introduction into reactor (2 in Fig. 12) to the moment of the explosion of the mixture.

Pressure time history (1 atm = 100 units); the valve circuit is (1) opened and (2) closed. Reproduced from [31] with permission from Pleiades Publishing, Ltd
The primary experimental data were reported in [31]. In Fig. 13, one can see the example of experimental results. It was found that, for systems containing activated nanodispersed powders, there exists a certain threshold temperature T th. At temperatures below the threshold value, the ignition delay time of the fuel suspensions turned out to be identical, within the measurement error, to that of n-heptane. At temperatures above the threshold value, the ignition delay time is much shorter, decreasing to 10−2 s. The lowest threshold temperature (277 °C) was found for the Mg/MoO3-containing system. For the Al/MoO3 system, the threshold temperature was 363 °C. For the Mg/PTFE-containing system, no threshold temperature was found up to 385 °C, while its ignition delay time was identical to that of heptane.

Temperature dependences of the ignition delay time of fuel–air mixture for heptane (squares) and heptane +2.5% (3 Mg + MoO3) (triangles). Reproduced from [31] with permission from Pleiades Publishing, Ltd
High-speed video recording data accomplished in Ref. [31] show that at start of injection of cold air in the nearby hot air, individual nanoparticles or small groups of them find themselves at hot reactor walls or in the nearby hot air, where they ignite. The ignition of these particles does not, however, ignite the system as whole. The kinetics of self-ignition of fuels with mechanoactivated additives at T ≥ T th is determined not so much by the kinetics of oxidation of the hydrocarbon component, as by the time of formation of combustible mixture and by the dynamic of heating of the hotspots due to the reaction of the conglomerates of activated nanoparticles.
To interpret these observations, the threshold self-ignition temperatures are superimposed in the form of arrows onto the curves of O2 release [37] during mechanoactivation of MoO3 (Fig. 8). As shown in Fig. 8, the self-ignition temperature coincides with the temperature range of oxygen release from the oxide.
Low-temperature stages of interaction of metals with oxygen take place via diffusion of ions through oxides layer of MgO and Al2O3, respectively. The rate of reaction determines the intensity of ions flow, which depends on the contact surface area. The contact surface area S C in the Mg/MoO3 system is significantly higher than that in the Al/MoO3 system (Table 1; Fig. 10), so the threshold temperature in the first case is lower than that it is in the second. It seems to us that the difference in S C is the most important factor for reaction acceleration. Nevertheless, in the future it is necessary to take into account the difference in the structure of Mg and Al, MgO and Al2O3, different energies, mechanism of reaction of oxidation, etc.
To understand why this effect is absent in the Mg/PTFE system, it is necessary to analyze the reactivity of this composition under slow heating.
Reactivity under conditions of slow heating
System Mg/(–C2F4–) n [34]
Mechanical activation initiates the chemical transformation in this system according to reaction (1)
The optimal dose of mechanoactivation at which the full conversion takes place is equal to 7–8 kJ/g [34].
Figure 14 shows the results of DSC measurements on Mg/(–C2F4–) n samples during their slow heating (10 °C/min). For the initial, inactivated mixture, an endothermic peak at 340 °C, associated with PTFE melting, is observed, but virtually no exothermic peaks are seen (curve 1). At temperatures above 470 °C, the sample weight decreases (curve 3) and gaseous products of PTFE depolymerization, C2F4 + molecular ion and its fragments (CF2 +, CF+, F+) are detected by mass spectrometer. After heating, the diffractograms showed no phase of MgF2, i.e., reaction (1) virtually does not occur. Thus, in the inactivated mixture, the depolymerization of (–C2F4–) n occurs, accompanied by the evolution of gaseous products, the interaction of which with the magnesium is negligible due to a low surface area of contact between the components.

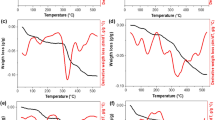
(1, 2) DSC and (3, 4) thermogravimetry curves for (1, 3) initial and (2, 4) mechanoactivated MAEC, 35/65 Mg/(–C2F4–) n
During heating of the activated mixture, which consists of magnesium particles encapsulated into PTFE, two exothermic effects at 400–500 °C were observed (curve 2), while the weight of the sample remained virtually unchanged (curve 4). After heating, diffraction methods showed that the solid products contain MgF2, but no magnesium and PTFE. Thus, in the activated mixture, reaction (1) occurred completely. The kinetics of accumulation of the product of reaction (1) with increasing temperature for samples with two different doses is displayed in Fig. 15.

Relative amount of MgF2 N = ([MgF2]/[MgF2(max)]) which is formed during heating of MAEC 35/65 Mg/(–C2F4–) n . D is the dose of MA
A comparison of the data in Figs. 14 and 15 suggests that there are three types of interaction of magnesium with PTFE in the activated mixtures. The main part of the chemical conversion takes place at temperatures above 420 °C. It is associated with the thermal depolymerization of the PTFE. For this channel, the thermal effect approximately corresponds to the enthalpy of conversion. Mechanoactivation shifts the depolymerization temperature by 50 °C (from 470 to 420 °C), whereas the core/shell structure of the composite and a high surface area of contact between the components in the activated mixture make it easy for gaseous depolymerization products to penetrate into the bulk of magnesium particles, thereby providing the completeness of reaction (1).
The second channel is implemented at lower temperatures (370–420 °C). The amount of MgF2 formed in this temperature range is ~5–7% (Fig. 15), whereas the thermal effect constitutes ~1.3% of the total thermal effect. Thus, the second channel is characterized by a low heat of conversion.
This result can evidently be explained by keeping in mind that the energy removal of the first fluorine atom C2F4 → C2F3+F is 134 kcal/mol, whereas the removal of the second atom, C2F3 → C2F2+F, requires an energy input of only 14 kcal/mol [46]. The ease of abstraction of the second fluorine atom, apparently, accounts for a large heat of reaction (1) when it proceeds through the PTFE depolymerization channel. The low-temperature reaction at 370–420 °C proceeds at the magnesium–PTFE interface, so it is reasonable to assume that, in the region of contact, only the first fluorine atom is abstracted from the polymer molecule and transferred to magnesium, which explains why the heat of reaction is low.
The third channel is realized at temperatures below 370 °C. In this temperature range, products in an amount of 10% were found, but no exothermic effects were detected. In addition, in this temperature range, the loss of dislocations and stalking faults was observed. So far, we can only assume that, in this case, the chemical reaction involves dislocations and packing defects migrated to the surface.
Thus, for MAEC Mg/(–C2F4–) n , the main role of MA is primarily to create a large surface area of contact between components and to form a core/shell structure. MA reduces the PTFE depolymerization temperature by ~50 °C, whereas the structure formed provides a complete interaction of the volatile depolymerization products with the metal. Various defects formed in the magnesium during MA are involved too in the formation of the final product of reaction (1). However, these processes are nearly energetically neutral and cannot act as initiating agents.
Mg/MoO3 system
In the case of Me/MoO3 MAEC, special attention was paid to the possibility of initiating a chemical transformation by oxygen released from activated MoO3 at low temperature.
Whether the oxygen released from the MoO3 participates in the oxidation of the metals was checked as follows: A mixture, composed of highly activated n–MoO3 and magnesium, was prepared by mixing the components in a mortar in a proportion corresponding to the stoichiometry of reaction (2). Then, the mixture was heated under DSC conditions. Along with the Mg and MoO3 phases, the products formed during DSC heating contained traces of the Mo4O11 and MgO phases were recorded. Thus, the diffraction method made it possible to confirm the transfer of oxygen from the oxidizer to the metal:
The results of experiments on the slow heating of activated Mg/MoO3 mixtures [35] are briefly summarized in Table 2. It was found that the nature of the interaction in the reaction depends on the sample weight and the oxygen pressure over the sample. For high vacuum, a relatively large sample (0.5 g) and a heating rate of ~5 °C/min, the onset of the explosive reaction mode occurred at 250 °C (run no. 1 in Table 2). We can assume that the initiation of the reaction is caused by the thermal decomposition of MoO3, with the beginning of oxygen evolution at this temperature. With a very low mass sample (2 mg) in DSC measurements (run no. 2 in Table 1), the reaction proceeds in one step at 470 °C. For a 20-fold larger sample, all other things being equal (run no. 3 in Table 1), the reaction during DSC measurements occur in the explosive mode. Note that, in these experiments, the inert gaseous medium (He) contained traces of oxygen. Finally, by increasing oxygen pressure under slow heating conditions (10 °C/min), it was possible to increase the temperature of reaction (2) (run no. 4 in Table 2) or even completely suppress it (run no. 5) by the competing reaction:
Thus, the lowest temperature at which the self-ignition of Mg/MoO3 MAEC occurs under conditions of slow heating is ~250 °C. At the same time, the self-ignition temperature is influenced by the sample weight, and the presence of oxygen in the gas phase, even in trace amounts: decreasing the sample weight and increasing the oxygen pressure, impedes the reaction.
Al/MoO3 system
There are numerous papers on measuring the thermal effects during heating mechanically activated composites Al/MoO3, see, e.g., Ref. [20,21,22,23]. Considerable attention was paid to a kinetic analysis of DSC curves and determination of kinetic parameters. In particular, it was reported [20] that, at temperatures of 200 to 240 °C, the activation energy and the frequency factor for the conversion reach their lowest values, with the conversion being described by a different kinetic law.
The results of our measurements are displayed in Fig. 16. The DSC curve also exhibits an exothermic effect at 150–300 °C, along with three broad exothermic peaks with maxima at 370–400, 500–580, and 750–850 °C, as well as a narrow endothermic peak of aluminum melting. Analysis of the solid products after partial heatings to 420, 680 and 1000 °C (Table 3) led us to conclude that the process involves the following reactions:
At 420 °C, the MoO2 phase appears, i.e., the first peak is due to a partial occurrence of reaction (4). By the time the temperature rises to 630–680 °C, the MoO3 turns out to be fully consumed, i.e., reaction (4) is completed, to give rise to the Mo phase, and reaction (5) begins. At the same time, the system still contains aluminum, which is confirmed by the diffraction data (Table 3) and the presence of the aluminum-melting peak in the DSC curves. The high-temperature peak in the DSC curves is due to the interaction of molten aluminum with MoO2 [reaction (5)], crystallization of aluminum oxides and interaction of excess aluminum with Mo to form MoAl12 MoAl8 or Mo3Al phases. The concurrent occurrence of a variety of processes explains the complex shape of the DSC curves. It may be proposed that thermal decomposition of activated MoO3 is the reason of low-temperature DSC peak.

DSC curves for heating mechanoactivated 40Al + 60MoO3 mixtures
Detonation-like processes
Investigations of the possibility of detonation-like processes in mechanically activated mixtures were carried out by initiating the reaction by detonation of high explosives. Studies have shown that the self-sustaining process rapidly damps in dense pressed mixtures and with strong initiation. But with a weak initiation in loose-packed mixtures, the propagation of a self-sustaining process with supersonic speed is entirely possible. Some results are shown in Fig. 17. The experiments were performed in metal or Plexiglas tubes 150–350 mm in height with an internal diameter of 10–30 mm. The test mixture powders were in portions filled in the tube and compacted to relative density of 0.25–0.38 theoretical maximum density. The process was initiated by a detonator with a small charge of RDX (hexogen) (1 g) and a bulk charge of 95/5 ammonium perchlorate/PMMA (polymethylmethacrylate) (10 g). Velocity was measured using electrical contact sensors, and, in some experiments, optical fibers were introduced into the studied mixture, along with the sensors, to record the light emission from the reaction products. At the end of the tube, there was a Duralumin witness plate, which allowed estimating the overall effect of the reacting system from the size of the crater at the center of the plate. In some experiments, composite plates of different materials with embedded PVDF (polyvinyldenfluoride) pressure gauges were placed at the end of the charge. In these experiments, detonation propagation at a constant velocity in low-density charges of activated Al/(–C2F4–) n and Mg/(–C2F4–) n was found. After initiation, a decaying process with an initial velocity of 1700–2000 m/s was recorded, followed by a stationary regime with velocities of 700–1300 m/s, depending on the composition and density of the charge. The velocity of the process greatly exceeded the sound speed in porous mixtures measured by an ultrasonic method (<100 m/s). The final velocity did not depend on the tube material and did not change when the diameter changes from 10 to 30 mm.

Experimental setup and velocities of propagation of the reaction initiated by a shock wave in some MAECs: 1—Al/(–C2F4–) n 27/73 D = 6 kJ/g, ρ 0 = 0.59 g/cc; 2—Al/(–C2F4–) n 45/55 D = 6 kJ/g, ρ 0 = 0.54 g/cc; 3—Al/(–C2F4–) n 25/75 D = 7.5 kJ/g, ρ 0 = 0.50 g/cc; 4—Mg/(–C2F4–) n 35/65 D = 8.8 kJ/g, ρ 0 = 0.49 g/cc; 5—Mg/(–C2F4–) n 45/55 D = 8.3 kJ/g, ρ 0 = 0.45 g/cc; 6—Al/MoO3 32/68 D = 3.18 kJ/g, ρ 0 = 1.43 g/cc; 7—Mg/MoO3 34/66 D = 2.9 kJ/g, ρ 0 = 1.13 g/cc
In similar experiments with Al/MoO3 and Mg/MoO3, a detonation process was not obtained. In the initial part of the charge, the process speed is maintained by the products of the initiating charge explosion, but then goes into a mode of explosive burning at speeds below 400 m/s.
From the point of view of the predominant role of the effective contact surface of reagents, it is evident that, for the Al/(–C2F4–) n and of Mg/(–C2F4–) n systems, the contact surface area is high enough, so that detonation-like modes with propagation velocities of more than 1 km/s can be realized. For the Mg/MoO3 system, with a sufficiently large surface area of contact between the components (Table 1), process propagates at a velocity of ~400 m/s, whereas for the Al/MoO3 systems, with the smallest S c , this mode was not implemented.
Discussion
For all the MAEC studied, mechanoactivation dramatically increases the surface area of contact between the components and produces various defects in both components. For both metals (Al and Mg), microstrain and small grain size were found. From the values of microstrain, the concentration of dislocation can be estimated. For Mg, the detailed analysis of the anisotropy of XRD lines broadening allows evaluation of the concentration of two types of stalking faults. In the activated MoO3 besides small grain size and microstrain, the “deformed” and “fractured” (paramagnetic centers Mo5+) Mo–O bonds were found. The parameters of these defects are collected in Table 1. Clearly, a large surface area of contact is an important condition for the occurrence of a chemical transformation. However, the focus was on the possible influence of defects in the components on the initiation of chemical conversion, in particular on the release of oxygen from activated MoO3.
Table 4 compares the temperature ranges of the processes considered in the present work. An important issue of thermal initiation is nucleation, so one can assume that, for the Al/MoO3 and Mg/MoO3 systems, spontaneous ignition occurs at temperatures at which oxygen is released from MoO3 (Fig. 8; Table 4). For the Mg/MoO3 system, the temperature of ignition in fuel–air mixtures is identical to the temperature of explosion in a vacuum of a large Mg/MoO3 sample. For the Mg/MoO3 composite, the threshold temperature is by ~100 °C lower than that for Al/MoO3. It is believed that this is due to a larger surface area of contact between the components in Mg/MoO3 as compared to Al/MoO3. For the Mg/PTFE system, the loss of defects in the magnesium component is not accompanied by an appreciable exothermic effect, although it leads to the beginning of the formation of MgF2, which explains why ignition does not occur. For small-weight samples slowly heated during DSC measurements, the situation is more complicated. It turned out that, for the Mg/MoO3 system, even a small addition of oxygen in the gaseous medium hinders the interaction with the lattice oxygen, possibly due to oxidation by oxygen from the gas phase. For the Al/MoO3 system, oxygen evolution from MoO3 or the rapid oxygen diffusion in the oxide, because of the presence of defects, presumably plays an important role. This effect possibly underlies the anomalous behavior of the kinetic parameters of the reaction at low temperatures (200–240 °C), which was observed in [23]. The oxygen release for metal/oxides systems was observed early in [47, 48].
In the shock-wave initiation of the reaction, the defects in the bulk and on the surface of the particles play a minor role. Therefore, detonation-like mode is realized only for systems Mg(Al)/PTFE with core/shell structure and large S C, whereas for the Mg/MoO3 velocity of propagation relatively small, and for Al/MoO3 this mode was not implemented.
Conclusion
The usual way of promotion of chemical transformation of energetic composites is increasing of contact surface area of components, first of all by decrease in particle size and using the nanoparticles. Under creation of energetic composites by methods of mechanical treatment, one additional possibility emerged to increase reactivity. Mechanical activation is accompanied by formation of metastable defects in components, which can be very active in chemical transformation. In the present paper, the results of our recent studies in this field are summarized. It is observed that the point defects in MoO3 initiate self-ignition process under fast heating of Me/MoO3 systems and can control the low-temperature reaction under the slow heating (DSC measurements). Of course, the detailed investigation of the reason for these phenomena will be carried out in the future. On the other hand, the reaction with defects in metals in Mg/PTFE system gives 10% of the reaction product (DSC measurements). But heat effect in this process is negligible, and there is no self-ignition in this system under fast heating. Under the conditions of shock-wave initiation, the presence of defects is non-important.
In conclusion, we would like to comment on the promise of energetic materials produced via mechanochemistry. The advantages of this method include improving properties (e.g., the rate of combustion or detonation) of energetic composites, the relative scalability, and comparatively low prices for preparation. The disadvantages are the danger of explosion under mechanical activation and following manipulation with activated composites, high sensitivity and worsening of the properties during the storage of activated composites. Some methods for overcoming of the disadvantages are developed now, and in our opinion, the mechanochemical methods of energetic materials preparation have promising future for using in special fields of industry.
References
Dreizin EL (2009) Metal-Based Reactive Nanomaterials. Prog Energy Combust Sci 35:141–167
Dixon GP, Martin JA, Thompson D (1998) Lead-free precussion primer mixes based on metastable interstitial composite (MIC) Technology, US Patent No. 5717159
Son SF, Busse JR, Asay BW, Peterson PD, Mang JT, Bockmon B, Pantoya ML (2002) Propagation studies of metastable intermolecular composites (MIC), in Twenty-Ninth International Pyrotechnics Seminar, Ed. by F. J. Schelling (IPS USA, Westminster), 203–212
Kolbanev IV, Butyagin PYu, Streletskii AN (2000) On the mechanochemistry of aluminum. Khim Fiz 19:96
Dolgoborodov AYu, Gogulya MF, Brazhnikov MA, Makhov MN, Fortov VE (2002) Detonation- like phenomena in Al/S Mixture, in Twenty-Ninth Int.Pyrotech. Seminar, Ed. by F. J. Schelling (IPS USA, Westminster, 2002), pp 557–563
Dolgoborodov AYu, Makhov MN, Gogulya MF, Streletskii AN, Kolbanev IV, Fortov VE (2003) Effect of Mechanical Activation on the Detonability of Oxidizer–Fuel Mixtures,” in V. E. Substances, Materials, and Structures under Intense Dynamic Loads, Ed. By A. L. Mikhailov (VNIIEF, Sarov, 2003), 273–278 [in Russian]
Suryanarayana C (2001) Mechanical alloying and milling. Prog Mater Sci 46:1–184
Grigor’eva TF, Barinova AP, Lyakhov NZ (2003) Mechanosynthesis of nanocomposites. J Nanoparticles Res 5:439–453
Grigor’eva TF, Korchagin MA, Barinova AP et al (1999) Influence of the mechanochemical activation on concentration limits of self- propagating high- temperature synthesis. Dokladi Akademii Nauk 369:345–347
Karagedov GR, Laykhov NZ (1997) Effect of mechanical activation on sintering of alumina. Inrganic Mater 33:688–691
Streletskii AN, Kolbanev IV, Borunova AB, Butyagin PYu (2004) Mechanochemically activated aluminium: preparation, structure and chemical properties. J Mater Sci 39:5175–5179
Pivkina A, Streletskii A, Kolbanev I, Ul’yanova P, Frolov Yu, Butyagin Yu, Schoonman J (2004) Mechanochemically activated nano-aluminium: oxidation behaviour. J Mater Sci 39:5451–5453
Streletskii AN, Kolbanev IV, Borunova AB, Yu Butyagin P (2005) Mechanochemical activation of aluminum: 3. Kinetics of interaction of Al with water. Colloid J 67:631–637
Streletskii AN, Mudretsova SN, Povstugar IV, Yu Butyagin P (2006) Mechanochemical activation of aluminum: 5. Formation of aluminum carbide upon heating of activated mixtures. Colloid J 68:623–631
Streletskii AN, Kolbanev IV, Borunova AB, Butyagin PYu (2010) Formation Mechanisms, Structure and Reactivity of “Mechanochemical” Nanocomposites”—In Experimental and Theoretical Studies in Modern Mechanochemistry, Ed. F. Delogu and G. Mulas, Transworld Research Network 37/661(2) Fort P.O. Kerala, India, pp. 169–189
Dolgoborodov AYu, Makhov MN, Kolbanev IV, Streletskii AN (2004). mechanically activated pyrotechnic composition, RF Patent No. 2235085, Buyl. Izobr. No. 24
Dolgoborodov AYu, Makhov MN, Streletskii AN, Kolbanev IV, Gogulya MF, Fortov VE (2004) On the possibility of detonation in a mechanically activated aluminum-polytetrafluoroethylene composite. Khim Fiz 23(9):85–88
Dolgoborodov AYu, Makhov MN, Kolbanev IV, Streletskii AN, Fortov VE (2005) Detonation in an aluminum-teflon mixture. JETP Lett 81:311–314
Dreizin EL, Schoenitz M (2009) Nano-composite energetic powders prepared by arrested reactive milling, US Patent No. 7524355 B2
Umbrajkar SM, Schoenitz M, Dreizin EL (2006) Control of structural refinement and composition in Al-MoO3 nanocomposites prepared by arrested reactive milling. Propellants Explos Pyrotech 31:282–289
Williams RA, Schoenitz M, Ermoline A, Dreizin EL (2014) Low-temperature exothermic reactions in fully-dense Al/MoO3 nanocomposite powders. Thermochim Acta 594:1–10
Sun J, Pantoya ML, Simona SL (2006) Dependence of size and size distribution on reactivity of aluminum nanoparticles in reactions with oxygen and MoO3. Thermochim Acta 444:117–127
Schoenitz M, Umbrajkar S, Dreizin EL (2007) Kinetics analysis of thermite reactions in Al-MoO3 nanocomposites”. J Propul Power 23:683–687
Dolgoborodov AYu, Streletskii AN, Makhov MN, Teselkin VA, Guseinov ShL, Storozhenko PA, Fortov VE (2012) Promising energetic materials composed of nanosilicon and solid oxidizers. Russ J Phys Chem B6:523–530
Dolgoborodov AYu, Ermolaev BS, Shevchenko AA, Teselkin VA, Kirilenko VG, Monogarov KA, Streletskii AN (2015) Combustion and detonation of mechanoactivated aluminum-potassium perchlorate mixtures. Russ J Phys Chem B 9:615–624
Peter Baláž, Marcela Achimovičová, Matej Baláž et al (2013) Hallmarks of mechanochemistry: from nanoparticles to technology. Chem Soc Rev 42:7571–7637
Dolgoborodov AYu (2015) Mechanically activated oxidizer-fuel energetic composites. Combus Explos Shock Waves 51:86–99
Dreizin EL, Schoenitz V (2015) Correlating ignition mechanisms of aluminum- based reactive materials with thermoanalytical measurements. Prog Energy Combust Sci 50:81–105
Rogachev AS, Mukasyan AS (2010) Combustion of heterogeneous nanostructural systems combustion. Explos Shock Waves 4:243–266
Energetic Nanomaterials: Characterization and Application. (2016) Ed. By Zarko V, 1st Edition, ISBN; 9780128027103, Elsevier
Troshin K Ya, Streletskii AN, Kolbanev IV, Borisov AA, Frolov SM, Frolov FS (2016) Promotion of the self-ignition of fuel-air mixtures with mechanoactivated Al(Mg)–MoO3. Russ J Phys Chem B10:435–443
Sterletskii AN, Dolgoborodov AYu, Kolbanev IV, Makhov MN, Lomaeva SF, Borunova AB, Fortov VE (2009) Structure of mechanically activated high-energy al + polytetrafluoroethylene nanocomposites. Colloid J 71:852–860
Streletskii AN, Kolbanev IV, Teselkin VA, Leonov AV, Mudretsova SN, Sivak MV, Dolgoborodov AYu (2015) Defective structure, plastic properties, and reactivity of mechanically activated magnesium. Rus J Phys Chem B9:148–156
Streletskii AN, Kolbanev IV, Leonov AV, Dolgoborodov AY, Vorobeva GA, Sivak MV, Permenov DG (2015) Defective structure and reactivity of mechanoactivated magnesium/fluoroplastic energy generating composites. Colloid J 77:213–225
Streletskii AN, Kolbanev IV, Ya Troshin K, Borisov AA, Leonov AV, Mudretsova SN, Artemov VV, Dolgoborodov AYu (2016) Structure and reactivity of mechanoactivated Mg(Al)/MoO3 nanocomposites. Russ J Phys Chem B 10:707–718
Sivak MV, Streletskii AN, Kolbanev IV, Leonov AV, Degtyarev EN, Permenov DG (2015) Defect structure of nanosized mechanically activated MoO3. Colloid J 77:333–340
Sivak MV, Streletskii AN, Kolbanev IV, Leonov AV, Degtyarev EN (2016) Thermal Relaxation of defects in nanosized mechanically activated MoO3. Colloid J 78:674–684
Streletskii AN (1993) Measurements and calculation of main parameters of power mechanical treatment in different mills. In: Proceedings of the 2nd International Conference on Structural Application of Mechanical Alloying. Vancouver, Canada, 20–22 sept. 1993. Ed. JJde Barbadillo, F H Froes, and R Schwarz, ASM International, Materials Park, Ohio, 44073-0002 P 51-58
Zakrevskiy VA, Tomashevskiy EE, Baptizmanskiy VV (1970) Visokolomecularnie soeginenija 12B:419 (in Russian)
Papakondylis A, Sautet P (1996) Ab Initio Study of the Structure of the α-MoO3 Solid and Study of the Adsorption of H2O and CO Molecules on Its (100) Surface. J Phys Chem 100:10681–10688
Py MA, Schmid PhE, Vallin JT (1977) Raman scattering and structural properties of MoO3. Il Nuovo Cimento 38:271–279
Mestl G, Srinivasan TKK, Knozinger H (1995) Mechanically Activated Moo3. 3. Characterization by vibrational spectroscopy. Langmuir 11:3035–3804
Mestl G, Verbruggen NFD, Knozinger H (1995) Mechanically activated MoO3. 2 Characterization of defect structures. Langmuir 11:3035–3041
Mestl G, Verbruggen NFD, Bosch E, Knozinger H (1996) Mechanically Activated MoO3. 5. Redox behavior. Langmuir 12:2961–2968
Dyrek K, Labanowska M (1991) Electron paramagnetic resonance investigation of the paramagnetic centres in polycrystalline MoO3. J Chem Soc Faraday Trans 87:1003–1009
Gurvich LV, Karachentsev GV, Kondrat’ev VN, Lebedev YuA, Medvedev VA, Potapov VK, Khodeev YuS, Energiya razryva khimicheskikhsvyazei. Potentsialy ionizatsii i srodstvo k elektronu (Energy of Chemical Bond Scission. Ionization Potentials and Electron Affinity), Moscow: Nauka, 1974
Williams RA, Shoenitz M, Ermoline A, Dreizin EL (2012) On gas release by thermally–initiated fully dense 2Al-3CuO nanocomposite powder. Int J Energ Mater Chem Propuls 11:275–292
Sullivan KT, Piekiel NW, Chowdhury S, Wu C, Zachariah MR, Johnson CE (2011) Ignition and combustion characteristic of nanoscale Al/AgIO3: a potential energetic biocidal system. Combust Sci Technol 183:285–302
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Project Nos. 16–03–00178a and 16–29–01030a) and the Program 14P of the Presidium of the Russian Academy of Sciences. The authors are grateful to V.V. Artemov and S.N. Lomaeva for providing unpublished microscopic data and center of magnet radio frequency spectroscopy of IBChPh RAS and E.N. Degtyarev for ESR measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Streletskii, A.N., Sivak, M.V. & Dolgoborodov, A.Y. Nature of high reactivity of metal/solid oxidizer nanocomposites prepared by mechanoactivation: a review. J Mater Sci 52, 11810–11825 (2017). https://doi.org/10.1007/s10853-017-1277-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1277-1