
Abstract
The volumetric energy density of today’s lithium-ion batteries is limited mostly by the graphitic carbon anode. Silicon is a promising replacement but its excessive volume expansion on lithiation limits its long-term cyclability performance. A nano-sized aluminium containing silicon, leached in acid, with a porous structure is shown to maintain its capacity higher than pure bulk silicon or nano-sized silicon by over 700 mAh/g. The capacity of leached silicon is maintained at 1400 mAh/g for more than 60 cycles. X-ray diffraction, scanning electron microscopy, transmission electron microscopy and nuclear magnetic resonance spectroscopy have been used to correlate the electrochemical performance with the materials' morphology and composition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There is an increasing demand for energy storage for a variety of portable and stationary applications including portable electronics, electric vehicles and grid smoothing and storage. Lithium-ion batteries, with a significantly higher energy density than aqueous nickel-based systems or lead acid batteries, have become the dominant storage medium [1]. Since their first commercialization, secondary lithium-ion batteries have used graphitic carbons as the active host for lithium at the anode because of its excellent cyclability. However, its capacity is only 0.8 Ah/cc, and takes up almost half the volume of today’s lithium-ion batteries. In addition, they operate at a potential close to that of metallic lithium, which introduces the possibility of lithium plating and dendrite formation with a catastrophic short circuit in the cell during cycling [2, 3].
A number of elements, such as aluminium, tin and silicon react rapidly with lithium under ambient conditions and are being considered as replacements for carbon. Each of these can store twice as much lithium as carbon on a volumetric basis, and generate much less heat and gas than carbon per lithium ion stored on thermal runaway and oxidation [4]. Electrochemical alloying of lithium with silicon forms alloys of Li x Si with x as high as 4.4 [2, 3], resulting in a much higher capacity than graphite (~4200 vs. 372 mAh/g). Moreover, the issue of solvent co-intercalation that can occur in graphitic carbons is circumvented in the case of Li–Si alloys [5–7]. However, the very high undesirable volume change on cycling leads to the decrepitation of the anode composite and necessitates the continuous rebuilding of the solid electrolyte interphase layer (SEI), leading to an exponential increase of cell resistance, material pulverization and loss of electrical contact between the active material and current collector [8]. Consequently, capacity fading and short battery life have been reported for observed Si bulk, Si thin films [9] and micron-sized particles [10]. The nanostructuring of silicon has been reported to improve the performance [11–14]. A number of nanoscale morphologies have been investigated to minimize electrode pulverization and subsequent cycling instability, such that nanostructured materials accommodate expansion and material stress by incorporating pores or voids, or by exploiting high surface-to-volume ratios [15]. Meanwhile, nanostructuring shortens lithium diffusion distances within the electrode, which benefits high rate capability [16]. For instance, Kohandehghan et al. reported that silicon nanowires (SiNWs) coated with magnesium and tin show significant improvement in electrochemical performance, the optimum architecture 3Sn/SiNWs maintained a reversible capacity almost double of baseline uncoated SiNWs [17, 18]. There is also some evidence that thin films of silicon with dimension of 1.2 µm prepared by chemical vapour deposition (CVD) obtains a discharge capacity of up to 4000 mAh/g over ten cycles, and evaporated thin films (40 nm) have shown stable capacities of up to 3000 mAh/g over 25 cycles [19, 20]. Nevertheless, as capacity is limited by cathode materials, exceedingly high capacity is not necessary for anodes, instead, long cycling life is indispensable. It has also been reported that Si nanotubes prepared by reductive decomposition of a silicon precursor in an alumina template and etching [21, 22], and Si nanowires realized using vapour–liquid–solid (VLS) or vapour–solid (VS) template-free growth methods [23], can both achieve the theoretical capacity of silicon with little fading during lithium insertion and removal process, but the improvement is limited, considering their tedious fabrication. Mechanical milling can significantly increase the specific surface area of the composite material [11, 24–26]. Graetz et al. reported that the cycle life depends on the nanostructured nature of the materials [26]. Introducing protection layers on the surface of silicon particles has been reported as an effective way to reduce side reaction and form stable SEI layers. Gaeun et al. reported that Si/Al2O3 foam anode with Al2O3-coated Si as the outer shell and Al2O3 as the core exhibits good cycling stability [27].
To address these issues, our research group has developed a new silicon anode with high loading and good electrochemical performance, which was derived from an Si–Al eutectic rather than from pure silicon [28]. The precursor eutectic contains 87.4 wt% Al and 12.6 wt% Si [29]. All but 5% of the Al can be removed by acid leaching. Most of this ~5 at.% of Al is dissolved as a solid solution in this nano-sized Si material. Here, we build on our earlier communication [29], determining the characteristics of this nano-sized porous aluminium containing silicon and comparing it with bulk silicon and nano-sized silicon.
Materials and methods
Three silicon materials were studied. A leached silicon was formed by etching a low-cost Al–Si alloy (325 mesh, 99%, Al:Si = 88:12 wt%, Alfa Aesar) in an excess of a 3 M aqueous solution of HCl for about 6 h, following our earlier procedure [ 28 ]. A second nano-Si was formed by milling bulk Si for 2 h, and a reference standard silicon from Umicore Company was provided by Lawrence Berkeley National Laboratory (LBNL).
The materials were characterized by synchrotron powder X-ray diffraction (XRD) on the beamline X14A at National Synchrotron Light Source with a wavelength of 0.7785 Å. The data were recorded during step scan in the 2θ range of 10°–60°. The Rietveld refinement of the XRD patterns was done using the GSAS/EXPGUI package [ 30, 31]. The morphology and elemental distribution were characterized by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS), on a zeiss supra field emission SEM operating at 10.0 and 15.0 kV, respectively. The electron diffraction and high angle annular dark field (HAADF) imaging in scanning transmission electron microscopy (STEM) imaging were carried out by transmission electron microscopy (TEM) JEOL JEM2100F with STEM and EDS elemental analysis. The chemical shift analysis was conducted by 29Si nuclear magnetic resonance spectroscopy (NMR) equipped with a Bruker AC 300 spectrometer with a Doty 7 mm CP/MAS probe. The 29Si frequency was 59.6 MHz under direct polarization experiment with an MAS spinning speed of 4 kHz, 5 μs 90° pulse, 4500–5500 scans and 15 s relaxation delay.
Electrochemical properties of these Si materials were evaluated in 2325-type coin cells under an argon atmosphere with lithium foil as the counter electrode, a Celgard 3501 separator, and a 1:1 volume mixture of ethylene carbonate and dimethyl carbonate (Novolyte) with 10% fluoroethylene carbonate additive were tested on a VMP2 multi-channel potentiostat (Biologic) at C/40 rate (current density 0.12 mA/cm2, ~48 mA/g) for the first cycle, followed by C/10 rate (0.5 mA/cm2, ~200 mA/g) for the remaining cycles from 2.0 to 0.01 V. The composites of the active material, carbon black (Cities Service Company) and sodium carboxymethyl cellulose (CMC, VMR) in a weight ratio of 70:20:10 were cast on copper foil, using an DI water slurry and dried at 85 °C in air for 12 h. Capacities were calculated based on the weight of Si (2–3 mg/cm2).
Results and discussion
The electrochemical behaviour of the silicon anode materials is shown for the first 60 cycles in Fig. 1. The leached Si material shows the most stable cycling, maintaining a capacity of ~1400 mAh/g over 60 cycles, whereas the standard Si although having the highest capacity over the first ten cycles fades rapidly. The milled Si material has the highest first cycle capacity of around 4800 mAh/g, which is over the theoretical capacity, but falls off very rapidly with a capacity of under 200 mAh/g at the 60th cycle. In the first cycle, some lithium ions are consumed to form SEI layer, and this amount of electron transfer is included in the first cycle capacity, so it is possible that the experimental capacity is greater than the theoretical capacity, especially for particles with dense defects and disorder structures. It is consistent with the morphological results shown in SEM figures, which will be discussed further in the following analysis. Based on the density of 0.96 g/cc for Li4.4Si and the capacity observed here of 1400 mAh/g, the calculated volumetric capacity of the leached Si is 1.3 Ah/cc after 60 cycles. This is significantly higher than the theoretical 0.75 Ah/cc for lithiated graphitic carbon, LiC6. For comparison, pure Li has a volumetric capacity of 2.1 Ah/cc. The capacity of the electrode is 980 mAh/g, which is still almost two times higher than graphitic carbon, the current anode in market.

Electrochemical data for Si electrodes. Capacity and efficiency versus cycle number, and voltage profiles for the first two galvanostatic cycles of a and b leached Si, c and d standard Si, and e and f milled Si. For all three materials, the first cycle was performed at 0.12 mA/cm2(C/40) and all subsequent cycles at 0.5 mA/cm2 (C/10)
All three materials showed a significant first cycle loss. The higher the first cycle capacity, 3300, 3700 and 4800 mAh/g, respectively for the leached, standard and milled silicon, the greater the loss in mAh/g. The Coulombic efficiency of the leached Si is 68% in the first cycle and increases to a steady 99% after a few cycles as indicated in Fig. 1a, which is higher than those of the other materials after the first few cycles. This initial irreversible capacity loss has been observed by others [23, 32], and can be attributed to surface reactions stemming from both SEI formation and lithiation of surface oxides. Although SEI formation has been observed for Si [33], we do not believe this to be the major cause of the initial excess capacity of any of the three silicon materials, because there is no appreciable capacity in the voltage range of the SEI formation (0.5–0.7 V) during the first cycle, [26] as observed in the voltage profiles in Fig. 1. Compared to the high charge capacity, the capacity involved in the SEI layer formation would be very small. Other side reactions with, for example, the added carbon may be possible causes, but the mechanism is not yet understood.
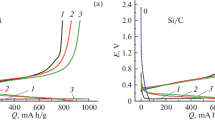
The leached silicon retains its high capacity even at high rates of reaction with lithium, as shown in Fig. 2. The capacity of leached Si at 0.15 mA/cm2 (C/15) is 1700 mAh/g, which is more than 50% higher than that of standard Si and 60% higher than that of milled Si both at the same C rate. Impressively, the capacity of leached Si at 2.3 mA/cm2 (C/1) still maintains 750 mAh/g, at which rate both standard Si and milled Si lose essentially all their capacity.

Capacity as a function of cycling rate for leached Si, standard Si and milled Si
The difference in electrochemical properties among the three silicon materials may be related to their different structures and morphologies. Accordingly, XRD measurements were carried out for the pristine powder of these materials. The XRD patterns with Rietveld refinement plots are shown in Fig. 3; and the detailed refinement results are summarized in Table 1, together with the crystallite size calculated using the Scherrer equation. All three pristine silicon materials show only diffraction peaks associated with Si. The standard silicon has the sharpest peaks, and this is at least in part due to the larger 81 nm crystallite size; it also has a lattice parameter identical to that of pure Si, 5.431 Å [34]. The leached silicon has a larger unit cell volume, which can be associated with the dissolution of some Al into the Si [28]. The unit cell volume of the milled silicon is again typical of that of pure silicon, and the broader peaks can be associated with the very small crystallite size, 20 nm. The nano dimension is likely due to the strain introduced by the milling.

XRD patterns and Rietveld refinement of a leached Si, b standard Si and c milled Si
The morphologies of these silicon materials were studied by SEM, as shown in Fig. 4. As reported by us [28] earlier the leached Si particles are spheres with dendritic flower-like structure, with each stem (diameter under 50 nm) packed side by side to form the outer layer. This morphology reveals that during solidification of the Al–Si eutectic, the Si dendrites grow before Al solidification and stop when they impinge on each other. After etching, almost complete absence of aluminium (∼88 wt% or 86 vol% in the original Al–Si) leaves a very porous structure inside the etched Si spheres [28]. The good performance of leached Si may be related to this porous structure: it is beneficial as a buffer media via leaving sufficient room for the particle’s expansion during lithium insertion, so as to enhance the capacity retention upon cycling the lithium in and out of the structure. In contrast, the other silicon materials do not have such an open morphology: standard Si exhibits a solid spherical morphology with a broad particle size distribution; the milled Si is aggregated into cube-like particles (~100 nm), also with a broad size distribution.

SEM images for a leached silicon material, b standard Si and c milled Si. The EDS results are also inserted into the images
Energy-dispersive X-ray spectroscopy mapping shows that there is around 5 wt% Al evenly distributed in the leached Si material (Fig. 4, inset), which is consistent with the XRD results. Overnight etching with vigorous stirring resulted in no change in the elemental distribution. The ~1 at.% Al in the standard Si (shown in EDS) might be an impurity introduced in the fabrication process, as the lack of a lattice parameter change suggests that it is not dissolved into the silicon. The microstructure of the leached Si along with its precursor AlSi alloy was investigated using TEM. The TEM images are shown in Fig. 5. The STEM high angle annular dark field (HAADF) image of leached Si (Fig. 5a) indicates dendritic particles, which are the broken parts of the flower-like silicon in Fig. 4. Figure 5b, c displays respectively an even distribution of Si, and just a trace of aluminium consistent with the EDS results. The precursor AlSi alloy shows an even distribution of both the Al and Si, and is consistent with around ~88% Al and 12% Si. The selected area electron diffraction patterns (Fig. 5g–i), exhibit well-defined spots indicative of predominantly crystalline Si in the leached material and predominantly Al in the precursor alloy.

TEM images of leached Si and AlSi alloy (precursor): a STEM HAADF image of leached Si; EDS elemental mapping of b Si and c Al in leached Si; d STEM HAADF image of AlSi alloy (precursor); EDS elemental mapping of e Si and f Al in AlSi alloy; the selected area electron diffraction patterns of g leached Si, h particle A and i particle B in AlSi alloy
Solid-state 29Si nuclear magnetic resonance (29Si NMR) spectra for the three Si materials and the precursor SiAl alloy shown in Fig. 6 indicate the characteristic peak of crystalline silicon, with a chemical shift of −81 ppm [35]. The lower broad peak seen in three of the materials at around −69 ppm can be associated with amorphous silicon [35, 36]. The signal seen around −115 ppm in the leached material is likely due to the presence of Si–O species formed during the acid leaching; the peak at −115 ppm is found in SiO2 [35].

29Si MAS NMR spectra for the three Si materials and the SiAl alloy
Conclusions
A rational control of morphology and composition plays a critical role in enhancing the electrochemical performance of Si as anode in lithium-ion batteries. A leached silicon material with its nano-sized crystallites and its nanoporosity shows enhanced capacity retention on cycling over both milled silicon and standard silicon particles. This may be associated with the openness of the morphology, which allows for the ready expansion of the lattice on reaction with lithium. These findings emphasize the importance of the control of crystallite size, porosity and chemical composition for silicon-based anodes for lithium batteries. This leached Si has a volumetric energy density 80% greater than that of graphitic carbon.
References
Winter M, Brodd RJ (2004) What are batteries, fuel cells, and supercapacitors? Chem Rev 104:4245–4269
Nesper R, Vonschnering HG (1987) Li2Si5, a zintl phase as well as a Hume-Rothery phase. J Solid State Chem 70:48–57
Weydanz WJ, Wohlfahrt-Mehrens M, Huggins RA (1999) A room temperature study of the binary lithium–silicon and the ternary lithium–chromium–silicon system for use in rechargeable lithium batteries. J Power Sour 81:237–242
Whittingham MS (2014) Ultimate limits to intercalation reactions for lithium batteries. Chem Rev 114:11414–11443
Besenhard JO, Yang J, Winter M (1997) Will advanced lithium-alloy anodes have a chance in lithium-ion batteries? J Power Sour 68:87–90
Winter M, Besenhard JO, Spahr ME, Novak P (1998) Insertion electrode materials for rechargeable lithium batteries. Adv Mater 10:725–763
Yazami R, Genies S (1998) Chemical stability of lithiated-HOPG with some organic electrolytes. Denki Kagaku 66:1293–1298
Winter M, Besenhard JO (1999) Electrochemical lithiation of tin and tin-based intermetallics and composites. Electrochim Acta 45:31–50
Baranchugov V, Markevich E, Pollak E, Salitra G, Aurbach D (2007) Amorphous silicon thin films as a high capacity anodes for Li-ion batteries in ionic liquid electrolytes. Electrochem Commun 9:796–800
Xie C, Lin ZL, Hanson L, Cui Y, Cui BX (2012) Intracellular recording of action potentials by nanopillar electroporation. Nat Nanotechnol 7:185–190
Li H, Huang XJ, Chen LQ, Wu ZG, Liang Y (1999) A high capacity nano-Si composite anode material for lithium rechargeable batteries. Electrochem Solid State Lett 2:547–549
Yu Y, Gu L, Zhu C, Tsukimoto S, van Aken PA, Maier J (2010) Reversible storage of lithium in silver-coated three-dimensional macroporous silicon. Adv Mater 22(20):2247–2250
Jia H, Gao P, Yang J, Wang J, Nuli Y, Yang Z (2011) Novel three-dimensional mesoporous silicon for high power lithium-ion battery anode material. Adv Energy Mater 1:1036–1039
Yao Y, McDowell MT, Ryu I, Wu H, Liu N, Hu L, Nix WD, Cui Y (2011) Interconnected silicon hollow nanospheres for lithium-ion battery anodes with long cycle life. Nano Lett 11:2949–2954
Bogart TD, Chockla AM, Korgel BA (2013) High capacity lithium ion battery anodes of silicon and germanium. Curr Opin Chem Eng 2:286–293
Szczech JR, Jin S (2011) Nanostructured silicon for high capacity lithium battery anodes. Energy Environ Sci 4:56–72
Kohandehghan A, Kalisvaart P, Kupsta M, Zahiri B, Amirkhiz BS, Li ZP, Memarzadeh EL, Bendersky LA, Mitlin D (2013) Magnesium and magnesium-silicide coated silicon nanowire composite anodes for lithium-ion batteries. J Mater Chem A 1:1600–1612
Kohandehghan A, Cui K, Kupsta M, Memarzadeh E, Kalisvaart P, Mitlin D (2014) Nanometer-scale Sn coatings improve the performance of silicon nanowire LIB anodes. J Mater Chem A 2:11261–11279
Sayama HYK, Kato Y, Matsuta S, Tarui H, Fujitani S (2002) In: Abstract 52, the 11th international meeting on lithium batteries, Monterey, CA
Takamura SOT, Suzuki J, Sekine K (2002) In: Abstract 257, the 11th international meeting on lithium batteries, Monterey, CA
Park MH, Kim MG, Joo J, Kim K, Kim J, Ahn S, Cui Y, Cho J (2009) Silicon nanotube battery anodes. Nano Lett 9:3844–3847
Lotfabad EM, Kalisvaart P, Kohandehghan A, Cui K, Kupsta M, Farbod B, Mitlin D (2014) Si nanotubes ALD coated with TiO2, TiN or Al2O3 as high performance lithium ion battery anodes. J Mater Chem A 2:2504–2516
Chan CK, Peng H, Liu G, McIlwrath K, Zhang XF, Huggins RA, Cui Y (2008) High-performance lithium battery anodes using silicon nanowires. Nat Nanotechnol 3:31–35
Wang CS, Wu GT, Zhang XB, Qi ZF, Li WZ (1998) Lithium insertion in carbon-silicon composite materials produced by mechanical milling. J Electrochem Soc 145:2751–2758
Kim I, Kumta PN, Blomgren GE (2000) Si/TiN nanocomposites-novel anode materials for Li-ion batteries. Electrochem Solid State Lett 3:493–496
Graetz J, Ahn CC, Yazami R, Fultz B (2003) Highly reversible lithium storage in nanostructured silicon. Electrochem Solid State Lett 6:A194–A197
Hwang G, Park H, Bok T, Choi S, Lee S, Hwang I, Choi NS, Seo K, Park S (2015) A high-performance nanoporous Si/Al2O3 foam lithium-ion battery anode fabricated by selective chemical etching of the Al–Si alloy and subsequent thermal oxidation. Chem Commun 51:4429–4432
Zhou W, Jiang T, Zhou H, Wang Y, Fang J, Whittingham MS (2013) The nanostructure of the Si–Al eutectic and its use in lithium batteries. MRS Commun 3:119–121
Murray AMJ (1984) The Al–Si (aluminum–silicon) system. J Phase Equilib 5:74–84
Larson AC, Von Dreele RB (2000) General structure analysis system (GSAS). Los Alamos National Laboratory report. LAUR 86-748, Los Alamos National Laboratory, Los Alamo
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34:210–213
Kasavajjula U, Wang C, Appleby AJ (2007) Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J Power Sour 163:1003–1039
Lee YM, Lee JY, Shim H-T, Lee JK, Park J-K (2007) SEI layer formation on amorphous Si thin electrode during precycling. J Electrochem Soc 154:A515–A519
Hull R (1999) Properties of crystalline silicon. IET, London
Jeong G, Kim Y-U, Krachkovskiy SA, Lee CK (2010) A nanostructured SiAl0.2O anode material for lithium batteries. Chem Mater 22:5570–5579
Hohl A, Wieder T, van Aken PA, Weirich TE, Denninger G, Vidal M, Oswald S, Deneke C, Mayer J, Fuess H (2003) An interface clusters mixture model for the structure of amorphous silicon monoxide (SiO). J Non-Cryst Solids 320:255–280
Acknowledgements
This research is based upon work supported by DOE-EERE, as part of BATT, DE-AC02-05CH11231 under Award Number 6807148. Use of the National Synchrotron Light Source at Brookhaven National Laboratory is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886. We thank Dr. Shailesh Upreti for providing the milled silicon sample. We also thank Qiyue Yin from Prof. Guangwen Zhou’s group for her help with TEM analysis, which was carried out in part at the Center for Functional Nanomaterials, Brookhaven National Laboratory, and supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-SC0012704. And we thank Dr. Juergen T. Schulte and Jordi Cabana for their help with the NMR analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jiang, T., Zhang, R., Yin, Q. et al. Morphology, composition and electrochemistry of a nano-porous silicon versus bulk silicon anode for lithium-ion batteries. J Mater Sci 52, 3670–3677 (2017). https://doi.org/10.1007/s10853-016-0599-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-0599-8