
Abstract
This work evaluated the thermal properties and biocompatibility of composites of polyhydroxyalkanoate (PHA) and hyaluronic acid (HA) (PHA/HA). Maleic anhydride (MA)-grafted polyhydroxyalkanoate (PHA-g-MA) was assessed as an alternative to PHA. The lower melt torque of the PHA-g-MA/HA composites resulted in them being more easily processed than PHA/HA. The water resistance of PHA-g-MA/HA was higher than that of PHA/HA. Human foreskin fibroblasts (FBs) were seeded on two series of these composites to assess cytocompatibility. Collagen and cell proliferation analyses indicated that PHA and PHA-g-MA and their composites were biocompatible with respect to FB proliferation. However, FB proliferation, collagen production, and the percentage of normal cells growing on PHA/HA composites were greater than those for PHA-g-MA/HA composites. Cell-cycle and apoptosis assays by FBs on the PHA series composite samples were not affected by DNA content related to damage; i.e., rapid apoptosis/necrosis was not observed, demonstrating the potential of PHA/HA or PHA-g-MA/HA membranes for biomedical material applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biomedical materials have found widespread application because of the development of medical technology and their improvement of the quality of life [1–3]. Biomedical material is a cross-disciplinary field, which includes biology, medicine, and materials science. Because biomedical materials can come into direct contact with human organs and blood, it is important to consider the stability, toxicity, biocompatibility, biodegradability, and other characteristics of these materials in the human body [4–6]. The study of biomedical materials has focused mainly on materials that can be decomposed biologically, such as polylactic acid (PLA), polycaprolactone (PCL), polyhydroxyalkanoate (PHA), and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) [7–10]. The addition of different materials to a composite can alter its processability, biocompatibility, and other characteristics, and provides an opportunity to optimize its performance [11, 12].
PHA is the biodegradable material currently drawing the most attention. It is a type of aliphatic polyester created mainly by the synthesis of β-polyhydroxyalkanoates [13, 14]. It is characterized by nontoxicity, biocompatibility, and good thermal processability. Given its good biocompatibility, PHA can be made into dental fillings, biological sutures, and fracture fixation materials. The absence of risk of toxic substances penetrating from it into the human body has led it to become commonplace in the biomedical materials market [15]. Composites are made in combination with hyaluronic acid (HA) to increase the functionality of PHA for biomedical engineering applications.
Hyaluronic acid has gradually become an important biomaterial. It is a transparent biopolysaccharide with a high molecular weight. Its structure is a linear biopolymer composed of d-glucuronic acid and N-acetyl-d-glucosamine repeated and alternated through the β-1,4 and β-1,3 glycoside bonds [16, 17]. Given its excellent biocompatibility, biodegradability, nontoxicity, and hypoallergenic reactivity, it can be widely distributed in the intercellular matrixes and extracellular matrixes (ECM) of human connective tissue, such as in the umbilical cord, synovial fluid, skin, vitreous humor, and serum [18, 19]. However, HA is a powder and is soluble in water; this makes it difficult to mold it into a film for biomedical applications. To address this issue, PHA (or PHA-g-MA)/HA biomedical film was synthesized.
This work investigated composite materials composed of PHA blended with HA as filler to enhance the utility of PHA in biomedical applications. To reduce the incompatibility between the HA and the PHA matrix, maleic anhydride (MA) was grafted to the PHA as an interface compatibilizing agent [20]. Bulk structural changes in the composites induced by the MA moiety were identified using Fourier transform infrared spectrometry (FTIR), 13C nuclear magnetic resonance (NMR) spectroscopy, and differential scanning calorimetry (DSC). Additionally, the biocompatibility of the composites was evaluated based on cell proliferation, collagen quantification, apoptosis, and the cell cycle.
Materials and methodology
Materials
Commercial-grade PHA (EM 5400F) was obtained from Shenzhen Ecomann Biotechnology (Shenzhen, China). Koch-Light Laboratories supplied the HA powder (MW = 1.7 × 106). MA, benzoyl peroxide (BPO), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was acquired from Promega (Madison, USA). Fetal bovine serum and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Life Technologies (Gibco–BRL; Gaithersburg, USA). Binding buffers and the Annexin V–FITC Apoptosis Detection Kit were obtained from BD Biosciences (Franklin Lakes, USA). All buffers and other reagents were of the highest purity grade commercially available.
PHA-g-MA copolymer
The grafting of MA onto molten PHA
In preliminary testing using 240 ± 1 mL of dichloromethane as the solvent, a mixture of 6.0 ± 0.2 g of MA and 1.8 ± 0.1 g of BPO was added as four equal doses at 2-min intervals to 54.0 ± 0.6 g of PHA. The grafting reactions were performed in a nitrogen (N2) atmosphere at 85 ± 2 °C. Preliminary results revealed that reaction equilibrium was attained in less than 12 h. Thus, reactions were allowed to progress for 12 h while stirring at 60 ± 1 rpm. The product (4.0 ± 0.2 g) was dissolved in 200 ± 2 mL of refluxing dichloromethane at 85 ± 2 °C, then the solution was filtered through several layers of cheesecloth and evaporated in a vacuum oven at 80.0 ± 1.0 °C over 24 h. The dichloromethane-insoluble product that remained on the cheesecloth was washed with acetone to remove any unreacted MA. The dichloromethane-soluble product in the filtrate was extracted using 600 mL of cold acetone.
The MA loading of the dichloromethane-soluble polymer (expressed as the grafting percentage) was calculated from the acid number and was determined as follows. First, 2.0 ± 0.1 g of copolymer was heated in 200 ± 1.0 mL of refluxing dichloromethane for 2 h. The hot solution was then titrated immediately with 0.03 ± 0.002 N ethanolic potassium hydroxide (KOH) solution, which was standardized against a solution of potassium hydrogen phthalate, using phenolphthalein as an indicator. The acid number was calculated using Eq. (1), and the grafting percentage was calculated using Eq. (2) [21].
The grafting percentage was found to be 1.05 wt% when the BPO and MA loadings were maintained at 0.3 and 10 wt%, respectively.
Composite preparation
The prepared HA powder samples were washed with acetone and dried in an oven at 105 °C for 24 h. Composites were prepared in a Plastograph 200-Nm mixer with a blade rotor (Model W50EHT, C. W. Brabender Instruments, South Hackensack, USA). The composites were mixed at 140–150 °C for 16 min at a rotor speed of 50 rpm. Samples were prepared with mass ratios of HA to PHA or to PHA-g-MA of 5:95, 10:90, 15:85, and 20:80. Residual MA in the PHA-g-MA reaction mixture was removed via acetone extraction prior to the preparation of PHA-g-MA/HA. After mixing, the composites were pressed into thin plates with a hot press and placed in a dryer for cooling. These thin plates were cut into standard sample dimensions for further characterization.
Characterization
Solid-state 13C NMR spectra were acquired with a Bruker AMX–400 NMR spectrometer. Spectra were obtained at 100 MHz under cross-polarization while spinning at the magic angle. Power-decoupling conditions were set with a 90° pulse and a 4-s cycle time. FTIR (Model FTS–7PC, Bio-Rad Laboratories, Hercules, USA) was used to investigate the grafting reaction of MA onto PHA and to verify bond formation between the anhydride carboxyl groups in PHA-g-MA and the –OH or –NH groups of HA. Samples subjected to FTIR analysis were first ground into fine powders in a milling machine, mixed with potassium bromide, and then pressed into pellets. Phase compatibility in the composites was evaluated using dynamic mechanical analysis (DMA) (Model 2080, TA Instruments, New Castle, USA) operating in film tension mode. Test specimens were rectangular thin films measuring 25 × 5 × 1 mm3. DMA tests were performed at a frequency of 1 Hz and a strain level of 0.075 % under conditions in which the temperature rose from 0 to 200 °C at a rate of 3 °C min−1. Static forces were selected from the experimental results in the linear elastic region in the absence of drawing effects. To specify this force, several stress–strain experiments were conducted beforehand, so that the ratio of the static force to the dynamic force could be kept constant during the measurements. The glass-transition temperature (T g), melting temperature (T m), and heat of fusion (ΔH f) were determined using a differential scanning calorimeter (DSC) (Model 2010, TA Instruments, New Castle, USA). The mass of the specimens ranged from 4 to 6 mg. Melting curves were recorded between 0 and 200 °C at a heating rate of 10 °C min−1. Values of T g, T m, and ΔH f were extracted from the temperatures and areas of the melting peaks in the DSC heating thermograms.
Water absorption
Samples were prepared for water absorption measurements by cutting them into 50 × 30-mm strips (thickness: 150 ± 3 μm), in accordance with ASTM Standard D570. The samples were dried in a vacuum oven at 50 ± 2 °C for 10 h, cooled in a desiccator, and then immediately weighed to the nearest milligram (this weight was designated W c). Following this, the samples were immersed in distilled water and maintained at 35 ± 2 °C for 30 days. During that time, they were removed from the water at 5-day intervals, gently blotted using tissue paper to remove excess water from their surfaces, immediately weighed to the nearest milligram (with this weight designated W w), and returned to the water. Each W w was an average value from three measurements. The percent weight increase due to water absorption (W f) was calculated to the nearest 0.01 %, according to Eq. (3):
Cell culture
Normal human foreskin fibroblasts (FBs) (WS-1, BCRC60300) were obtained from the Bioresource Collection and Research Centre in Taiwan. The WS-1 cells were grown in a culture medium containing 90 % DMEM with 2 mM l-glutamine and Earle’s BSS adjusted to include 1.5 g L−1 of sodium bicarbonate, 0.1 mM nonessential amino acids, and 1.0 mM sodium pyruvate with 10 % (v/v) fetal bovine serum. The cells were cultivated at 37 °C in a humidified incubator in a 5 % CO2 atmosphere.
Cell proliferation assay
MTT assays were used to evaluate the proliferation of WS-1 cells on the membranes. Sample sheets were placed in 24-well plates and sterilized by exposure to ultraviolet light for 1 h. Each well plate was embedded with 2–4 × 104 cells, which were incubated in an atmosphere of 5 % CO2 at 37 °C for 1, 3, and 7 days. At the end of the incubation period, the culture medium in the plates was discarded, and the cells were washed with phosphate-buffered saline. Each well plate was then filled with 100 μL of MTT and placed in a dark environment. The culture was incubated for 5 h to convert the MTT to formazan. The supernatant was then removed, and 150 μL of DMSO was added to dissolve the formazan. The plates were shaken and stirred to ensure a uniform solution. The optical density at 540 nm was determined using a PowerWave XS reader (Bio-Tek Instruments, Winooski, USA).
Collagen quantification assay
Aliquots of the culture medium (0.1 mL) from the samples and Sircol dye reagent (1 mL) were mixed in a centrifuge tube for 1 h. The mixture was then centrifuged at 12000×g for 10 min, and the supernatant was removed. Any residual liquid clinging to the centrifuge tube was removed with absorbent paper. Care was taken not to touch the precipitate during this process. A 100-μL aliquot of 0.1 M NaOH was then added, and the sample was left for 15 min to dissolve the precipitate. After 10 min of stable coloration, 0.2 mL of the resulting liquid was removed and placed into a 96-well plate. The absorbance of the solution in the well was measured at 540 nm.
Determination of the cell-cycle progression
To measure the effect of the material on the cell cycle of a cell strain, the distribution of the cell cycle was analyzed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, USA) with propidium iodide (PI) dye. Composite (PHA or PHA-g-MA series) specimens were loaded in 6-cm culture dishes and sterilized by ultraviolet light for 60 min. Each dish was given 3 × 105 WS-1 cells for culture, with the exception of the control group. After 24 h of culture, the cells were collected and fixed by 70 % ethanol for at least 8 h to rupture the membrane, and the supernatant was removed by centrifugation. The PI/Triton X–100 DNA staining solution (0.2 mL containing 1 mg mL−1 PI (BD Biosciences), 0.2 mL containing 5 % Triton X–100 (Sigma), and 1.0 mL containing 2 mg mL−1 RNase A (Sigma) to 8.6 mL PBS (final volume: 10 mL)) was added. Cell DNA was measured using RNase to remove the RNA because PI combined with RNA during testing. After 30 min of reaction in a dark environment at room temperature, the flow cytometer and FL2 PMT were used to analyze 1 × 104 cells. The distribution of cells in the cell cycle was determined using WinMDI ver. 2.8 (The Scripps Research Institute, USA).
Apoptosis analysis
Aliquots of 1 × 106 cells were placed into small centrifuge tubes. Cells were spun at 200 g for 5 min, the supernatant was discarded, and the pellets were resuspended in 500 μL of phosphate-buffered saline containing 5 μL of Annexin V–FITC and 5 μL of the PI labeling solution. The cell suspension was incubated for 15 min at room temperature in the dark. Phosphate-buffered saline (500 μL) was added, and the stained cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences.). Data were analyzed using WinMDI ver. 2.8.
Statistical analysis
All experiments were repeated three times. The data are expressed as mean ± standard error (SE). Data were analyzed using the IBM SPSS Statistics (ver. 22) software, by one-way analyzed of variance (ANOVA) and t tests, with significant difference determined at the level of p < 0.05.
Results and discussion
Structural analysis of PHA and of PHA-g-MA and its composites
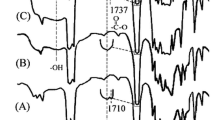
FTIR spectroscopy was used to investigate the grafting of MA onto PHA. The FTIR spectra of PHA and PHA-g-MA are shown in Fig. 1a, b. All of the characteristic peaks of PHA at 3200–3700, 1700–1760, and 500–1500 cm−1 appeared in both polymers [22], but two extra peaks were observed for the PHA-g-MA at 1785 and 1851 cm−1. These features are characteristic of MA groups; similar results have been reported previously [23]. The shoulders in the spectra represent the anhydride carboxyl groups in the modified polymer, signifying grafting of MA onto PHA. The FTIR spectra of PHA/HA(10 wt%) and PHA-g-MA/HA(10 wt%) are shown in Fig. 1c, d, respectively. A comparison of the spectra of PHA/HA (10 wt%) and PHA showed that the peaks assigned to –NH and –OH bond stretching vibrations at 3200–3700 cm−1 were considerably more intense for the PHA/HA (10 wt%) blend. This is because of contributions from the –NH and –OH groups of HA [24]. Expansion of the FTIR spectra in the limited range of 1700–1760 cm−1 (Fig. 1) more clearly illustrates the difference between the PHA/HA and PHA-g-MA/HA spectra. Figure 1a shows that for the PHA, the –C = O stretching vibration appeared as a strong broad band at 1700–1760 cm−1. The PHA/HA(10 wt%) blend displayed a broad band at 1700–1760 cm−1 (Fig. 1c), while PHA-g-MA/HA exhibited two new peaks at 1717 and 1737 cm−1 (Fig. 1d). The peak at 1717 cm−1 was assigned to the –C = O stretching vibration of the –N(COR)2 function, while the peak at 1737 cm−1 was attributed to the –C = O stretching absorption of the –OCOR function. These ester and imide functional groups formed via the reaction between the anhydride carboxyl groups of the PHA-g-MA and the –NH or –OH groups of HA when the two polymers were blended.

FTIR spectra (a) PHA, (b) PHA-g-MA, (c) PHA/HA (10 wt%), (d) PHA-g-MA/HA (10 wt%)
The solid-state 13C NMR spectra of PHA and PHA-g-MA were measured to further confirm this finding; these spectra are shown in Fig. 2a, b, respectively. Three peaks were observed, corresponding to carbon atoms in the unmodified PHA (1, 6: = 169.1; 2: δ = 38.7; 3: δ = 71.9; 4: δ = 26.5; 5: δ = 9.3; 7: δ = 40.6; 8: δ = 67.5; 9: δ = 19.7 ppm) [25]. The 13C NMR spectrum of PHA-g-MA showed additional peaks (10: δ = 42.1; 11: δ = 37.3; 12: δ = 171.3 ppm), thereby confirming that MA was covalently grafted onto the PHA.

Solid-state 13C NMR spectra a PHA, b PHA-g-MA, c PHA/HA (10 wt%), d PHA-g-MA/HA (10 wt%), e HA
The solid-state 13C NMR spectra of PHA/HA (10 wt%), PHA-g-MA/HA (10 wt%), and free HA are shown in Fig. 2c–e. The spectrum of free HA is in good agreement with that reported by Lopes et al. [26]. Comparing the PHA/HA (10 wt%) and PHA-g-MA/HA (10 wt%) spectra, we identified extra peaks at δ = 42.1 (10) and δ = 37.3 ppm (11) in the latter. As already indicated, the peaks at locations 10 and 11 for PHA-g-MA were from MA grafted onto PHA. The peak at δ = 171.3 ppm corresponding to the –C = O carbon of PHA-g-MA (Fig. 2b) was absent in the solid-state spectrum of PHA-g-MA/HA (10 wt%) (Fig. 2d); there appeared instead four peaks (13: δ = 173.2; 14: 176.2; 15: 178.1; 16: 179.3 ppm). These four peaks are evidence of the condensation reaction between the anhydride carboxyl groups of PHA-g-MA and the –OH or –NH groups of HA. Figure 2c shows that this reaction did not occur with PHA and HA.
Thermal properties of PHA and of PHA-g-MA and its composites
Figure 3 shows the effects of HA content and mixing time on the melt torque of the PHA/HA and PHA-g-MA/HA composites. When preparing PHA/HA or PHA-g-MA/HA, the polymer was first melted and HA was added to the melt. Therefore, the resulting polymer composite contained powder filler. The torque decreased with increasing HA content and mixing time, and leveled-off after 8 min of mixing. The final torque decreased with increasing HA content because the melt torque of HA was lower than that of PHA or PHA-g-MA. Additionally, because of the soft ester groups formed by reaction of PLA-g-MA and HA, the melt torque of the PHA-g-MA/HA was significantly lower than that of the PHA/HA for a given HA content.

Torque values as a function of mixing time for PHA/HA and PHA-g-MA/HA composites with various levels of HA
The dynamic mechanical properties of the PHA/HA and PHA-g-MA/HA composites were measured and used to evaluate the compatibility of hybrids. Figure 4a–c shows the variation of the storage modulus (E′), the loss tensile modulus (E″), and the loss tangent (tan δ) as a function of temperature for the neat PHA/HA and PHA-g-MA/HA composites. Regardless of the HA content, E′ (Fig. 4a) decreased while E″ (Fig. 4b) increased with increasing temperature and there existed a transition at ca. –2 to 5 °C for E″. The above phenomena may be related to the T g of the PHA, which occurs at this temperature. At the T g, the heat of deformation and molecular motions lead to energy dissipation. Note that the temperature dependences of E′ and E″ for the PHA-g-MA/HA composites were lower than those for the PHA/HA composites; i.e., E′ and E″ of the grafted polymer blends remained relatively high over a wide temperature range. This was attributed to the formation of ester and imide bonds.

Dynamic viscoelastic behavior of PHA/HA and PHA-g-MA/HA composites containing various levels of HA
Figure 4(c) shows that tan δ increased sharply at a particular temperature, which corresponded to the onset of segmental motion. The temperature of maximum damping shifted to higher values with increasing HA content because of the underlying influence of the T g of the amorphous phase in the semi-crystalline PHA-g-MA. In the presence of 10 wt% of HA, the peak damping temperature had shifted by ca. 8.5 °C. The shift resulted from enhanced restriction of the motion of the amorphous phase, suggesting increased adhesion between the polymer and HA in these composites. In the case of the PHA/HA composites, the tan δ curve generally broadened and decreased in intensity with increasing HA content. It is clear that the damping temperature of PHA/HA composites only slightly increased (by ca. 4.5 °C) as the HA content increased from 0 to 10 wt%, suggesting a smaller enhancement in the restricted motion of the amorphous phase of the PHA/HA composites; this relates to the poor compatibility between the HA and PHA phases. Furthermore, these results indicated only a small enhancement in the restricted motion of the amorphous phase and a decrease in the chain mobility of the PHA-g-MA/HA composites because of the poor compatibility between the PHA and HA.
The ΔH f, T m, and T g of the PHA/HA and PHA-g-MA/HA composites with different HA contents were determined using DSC. The results are shown in Table 1. For both composites, the T m decreased with increasing HA content, reflected by the aforementioned decreasing melt torque [27]. For the same HA content, the PHA/HA composite had a higher T m than did the PHA-g-MA/HA composite. This trend is consistent with the corresponding torque measurements shown in Fig. 3. The lower melt torque suggests that PHA-g-MA/HA would be more processable than PHA/HA. On the other hand, the T g increased with increasing HA content for both PHA/HA and PHA-g-MA/HA. This increase is likely a result of the reduced space available for molecular motion with increasing HA content in the composites [28]. The T g values were higher for the PHA-g-MA composites by approximately 6–12 °C.
The ΔH f of pure PHA and PHA-g-MA were 41.5 and 39.9 J g−1, respectively. The lower ΔHf of PHA-g-MA is attributed to grafted branches that disrupt the regularity of the chain structures in PHA and increase the spacing between polymer chains. The values of ΔHf for PHA-g-MA/HA were ca. 3–10 J g−1 higher than those of PHA/HA, probably because of condensation reactions, as discussed above. ΔH f may be used as an indicator of blend crystallinity. Although the ΔH f of both the PHA/HA and PHA-g-MA/HA blends decreased with increasing HA content (Table 1), the extent of the decrease was significantly greater in PHA/HA, indicating a lower degree of crystallinity. These results are similar to those obtained elsewhere with composites of polymers and natural fibers [29].
Water absorption of PHA and of PHA-g-MA and its composites
For the same HA content, the PHA-g-MA/HA composites had greater resistance to water absorption than did the PHA/HA composites (Fig. 5). The water resistance of the PHA-g-MA/HA composites was moderate, and the hydrophobicity of HA was probably enhanced by interactions with PHA-g-MA. For PHA/HA and PHA-g-MA/HA, the percent weight gain over the 15-day test period increased with the HA content. Since the arrangement of polymer chains in these systems was supposedly random, this result was likely due to decreased chain mobility with greater amounts of HA and to the hydrophilic character of HA, which adhered weakly to the more hydrophobic PHA.

Percent weight gain from water absorption of the PHA/HA and PHA-g-MA/HA composites is shown as a function of time
Cell proliferation of PHA and of PHA-g-MA and its composites
The biocompatibility of the composites was evaluated by measuring the cell growth rate of normal FBs seeded on the membranes (Fig. 6a). Cell viability was examined using MTT assays. The FB cell growth on the membranes was measured on days 1, 3, and 7. Seeding onto the PHA/HA membranes yielded similar cell viabilities from day 1 to day 7, indicating good biocompatibility with FB cells. Cell viability did not exhibit a decreasing trend as a function of increasing HA content; similar results were obtained at HA contents ranging from 5 to 15 wt% (data not shown). The cell viability of the PHA-g-MA membranes was significantly different from that of the control at day 7 (Fig. 6); this suggests that MA had a slight cytotoxic effect on the cells.

a Cell proliferation ratios of human FBs seeded on two series of membranes. From 1 to 7 days, the cell proliferation rate was quantified via an MTT assay. b The amount of HA secreted from human foreskin FBs seeded on two series membranes for 7 days. Sircol dye was used to quantify the amount of HA. c SEM images of human foreskin FBs and HA on PHA, PHA-MA, PHA/HA, and PHA-g-MA/HA membranes after 7 days of culture (scale bar 20 μm). All data are expressed as mean ± standard error (SE) of at least three separate experiments. Statistical analysis was performed using t tests, with significant differences asterisk determined at the level of p < 0.05 for each treatment versus the control group
Collagen production of PHA and of PHA-g-MA and its composites
Sirius red dye was used to stain the collagen secreted by dermal FBs in the wells of the plate reader or on the composite membranes (Fig. 6b). Collagen production did not vary significantly between the treatments and the control (p < 0.05), indicating that the composite materials were biocompatible with the cells. However, although not significantly different, collagen production was lower for the PHA-g-MA composite treatment versus the control at day 7, in addition to a reduction in cell viability; these findings suggested that MA had an intrinsic cytotoxic effect on the cells.
Collagen is an important component for cell proliferation and tissue formation, both of which are dependent on FB production [30, 31]. Therefore, the concentration of secreted collagen is an important indicator of successful wound healing. Because the PHA/HA membranes stimulated collagen secretion by FB cells, they may be suitable biomaterials for tissue engineering.
Figure 6c shows SEM images of collagen and FBs on the membranes after 7 days of culture. Thread-like collagen formations can be seen on the membranes. These SEM data show the compacted fibril morphology of collagen. Increasing levels of collagen secretion became evident in the SEM images as the percentage of HA in the PHA/HA composites increased. This is consistent with the collagen quantification data (Fig. 6b) and suggests that these membranes stimulate collagen production.
Cell-cycle assays of PHA and of PHA-g-MA and its composites
In the cell-cycle test (Fig. 7), the horizontal coordinate (FL2–A) corresponds to the fluorescence peak area, which reflects the cellular DNA content; the vertical coordinate (or event) shows the number of cells. In Fig. 6a, the first absorption peak represents the cell during the DNA growth period (G1); the second absorption peak represents the DNA of the cell in the mitosis period (G2/M); and the region between the G1 and G2/M absorption peaks represents the DNA of the cell in the synthesis phase (S). Saxena et al. [32] used a flow cytometer to measure the DNA of human cells and analyzed the cell cycle in G1, S, and G2/M phases to evaluate the relationship between the cell cycle and apoptosis.

a The horizontal axis (FL2–A) corresponds to the cell DNA content. The vertical axis (event) represents the cell number. b Cell-cycle histograms represent the percentage of the total cell population in the different phases. All data are expressed as mean ± SE of at least three separate experiments. Statistical analysis was performed using t tests, with significant differences asterisk determined at the level of p < 0.05 for each treatment versus the control group
Figure 7a shows the cell-cycle results (Fig. 7a) for various samples cultured with WS-1 cells for 24 h. The cell cycles were compared with those of the control group; a slight difference (p < 0.05) was evident between the cell cycles in close G1, S, and G2/M absorption positions. The DNA content area changed slightly. A slight difference (p < 0.05) in the percentage of cells was observed between the G1, S, and G2/M phases of various sample composites (Fig. 7b). No significant effect on the cell cycle of the WS-1 cell strains was observed among various sample materials. The above results indicate that PHA/HA and PHA-g-MA/HA composites do not damage cells.
Apoptosis assays of PHA and of PHA-g-MA and its composites
The nucleic acid stain PI and Annexin V–FITC (ApopNexin FITC Apoptosis Detection Kit, Chemicon, Millipore, Billerica, USA) were used to double-stain WS-1 cells. Stained cells were quantified using a flow cytometer (FACSCalibur, BD Biosciences), which allowed quantitative differentiation of normal cells, early- and late-stage (total) apoptosis cells, and necrotic cells [33, 34].
Figure 8 shows the populations of normal-, early-, and late-stage total apoptotic and necrotic WS-1 cells on PHA/HA and PHA-g-MA/HA composites. The population of normal cells for each treatment was not significantly different (p < 0.05) from that of the control group. However, the population of normal cells on the PHA/HA composites tended to be slightly greater than that on PHA alone or on PHA-g-MA/HA composites. This finding supports the above biocompatibility results, demonstrating that the addition of HA enhanced the biocompatibility of the final composite and is therefore unlikely to induce cell necrosis or apoptosis. Conversely, the cell populations on the PHA alone or PHA-g-MA/HA composite contained greater proportions of necrotic and apoptosis cells (p < 0.05). These results were attributed to the presence of MA and HA; MA induced a slight cytotoxicity, and HA enhanced the biocompatibility. Despite the lack of a statistically significant difference, the normal cell population on the PHA/HA composites was 2–4 % larger than that on the PHA-g-MA/HA composites.

a The horizontal axis (FL1–H) represents the cells stained by annexin V-FITC. The vertical axis (FL2–H) represents the cells stained by PI. b Apoptosis histograms showing the percentage of the total cell population in the different phases. All data are expressed as mean ± SE of at least three separate experiments. Statistical analysis was performed using t tests, with significant differences asterisk determined at the level of p < 0.05 for each treatment versus the control group
Conclusions
The structural, thermal, and biological properties of PHA/HA and PHA-g-MA/HA composites were investigated. Condensation reactions between HA and MA groups in PHA-g-MA occurred, as revealed by FTIR and 13C NMR analyses, and resulted in significantly different structures in the two composite materials. Although DSC tests revealed a decrease in the melting temperatures of both PHA/HA and PHA-g-MA/HA with increasing HA content, the PHA-g-MA/HA composites were more processable because of their lower melting temperatures and mixing torques. The T g of PHA-g-MA/HA was higher than that of PHA/HA, indicating more hindered molecular motion. The water resistance of PHA-g-MA/HA was higher than that of PHA/HA. Collagen and cell proliferation analyses indicated that the PHA or PHA-g-MA and their composites were biocompatible with respect to FB proliferation. The cell-cycle and apoptosis assays by FBs on the PHA series composite samples demonstrated the potential of PHA/HA or PHA-g-MA/HA membranes for biomedical material applications. The cytocompatibility of PHA-g-MA/HA was lower than that of PHA/HA.
References
Camci-Unal G et al (2013) Synthesis and characterization of hybrid hyaluronic acid-gelatin hydrogels. Biomacromolecules 14(4):1085–1092
Schanté CE et al (2013) Chemical modifications of hyaluronic acid for the synthesis of derivatives for a broad range of biomedical applications. Carbohydr Polym 85(3):469–489
Jeong CG et al (2014) Screening of hyaluronic acid–poly(ethylene glycol) composite hydrogels to support intervertebral disc cell biosynthesis using artificial neural network analysis. Acta Biomater 10(8):3421–3430
Bauer S et al (2013) Engineering biocompatible implant surfaces Part I: materials and surfaces. Prog Mater Sci 58(3):261–326
Waldby C et al (2004) Blood and bioidentity: ideas about self, boundaries and risk among blood donors and people living with Hepatitis. Soc Sci Med 59(7):1461–1471
Stamatialis DF et al (2008) Medical applications of membranes: drug delivery, artificial organs and tissue engineering. J Membr Sci 308(1–2):1–34
Volova T et al (2003) Results of biomedical investigations of PHB and PHB/PHV fibers. Biochem Eng J 16(2):125–133
Tanase CE, Spiridon I (2014) PLA/chitosan/keratin composites for biomedical applications. Mater Sci Eng C 40:242–247
Serrano MC et al (2004) In vitro biocompatibility assessment of poly(ε-caprolactone) films using L929 mouse fibroblasts. Biomaterials 25(25):5603–5611
Yang X, Zhao K, Chen G-Q (2002) Effect of surface treatment on the biocompatibility of microbial polyhydroxyalkanoates. Biomaterials 23(5):1391–1397
Wang T-J et al (2012) The phenotypic response of bovine corneal endothelial cells on chitosan/polycaprolactone blends. Colloid Surface B 90:236–243
Zhang S et al (2013) Novel toughening mechanism for polylactic acid (PLA)/starch blends with layer-like microstructure via pressure-induced flow (PIF) processing. Mater Lett 98:238–241
Laycock B et al (2013) The chemomechanical properties of microbial polyhydroxyalkanoates. Prog Polym Sci 38(3–4):536–583
Hazer DB, Kılıçay E, Hazer B (2012) Poly(3-hydroxyalkanoate)s: diversification and biomedical applications A state of the art review. Mater Sci Eng, C 32:637–647
Chen G-Q, Wu Q (2005) The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials 26(33):6565–6578
Luo Y, Prestwich GD (2001) Hyaluronic acid-N-hydroxysuccinimide: a useful intermediate for bioconjugation. Bioconjugate Chem 12(6):1085–1088
El-Safory NS, Fazary AE, Lee C-K (2010) Hyaluronidases, a group of glycosidases: current and future perspectives. Carbohydr Polym 81(2):165–181
Lai J-Y, Tu I-H (2012) Adhesion, phenotypic expression, and biosynthetic capacity of corneal keratocytes on surfaces coated with hyaluronic acid of different molecular weights. Acta Biomater 8(3):1068–1079
Levett PA et al (2014) A biomimetic extracellular matrix for cartilage tissue engineering centered on photocurable gelatin, hyaluronic acid and chondroitin sulfate. Acta Biomater 10(1):214–223
Quiroz-Castillo JM et al (2014) Preparation of extruded polyethylene/chitosan blends compatibilized with polyethylene-graft-maleic anhydride. Carbohydr Polym 101:1094–1100
Srubar WV et al (2012) Mechanisms and impact of fiber–matrix compatibilization techniques on the material characterization of PHBV/oak wood flour engineered biobased composites. Compos Sci Technol 72(6):708–715
Haynes D et al (2007) In Situ Copolyesters Containing Poly(l-lactide) and Poly(hydroxyalkanoate) Units. Biomacromolecules 8(4):1131–1137
Yu T, Jiang N, Li Y (2014) Study on short ramie fiber/poly(lactic acid) composites compatibilized by maleic anhydride. Compos Part A 64:139–146
Alkrad JA et al (2003) Characterization of enzymatically digested hyaluronic acid using NMR, Raman, IR, and UV–Vis spectroscopies. J Pharm Biomed Anal 31(3):545–550
Chuah J-A et al (2010) Biosynthesis and characterization of polyhydroxyalkanoate containing 5-hydroxyvalerate units: effects of 5HV units on biodegradability, cytotoxicity, mechanical and thermal properties. Polym Degrad Stabil 95(12):2226–2232
Lopes TD et al (2014) Bacterial cellulose and hyaluronic acid hybrid membranes: production and characterization. Int J Biol Macromol 67:401–408
Lei Y et al (2007) Preparation and properties of recycled HDPE/natural fiber composites. Compos Part A 38(7):1664–1674
Sanchez-Garcia MD et al (2008) Morphology and barrier properties of solvent cast composites of thermoplastic biopolymers and purified cellulose fibers. Carbohydr Polym 71(2):235–244
Beg MDH, Pickering KL (2008) Accelerated weathering of unbleached and bleached Kraft wood fibre reinforced polypropylene composites. Polym Degrad Stabil 93(10):1939–1946
Arahira T, Todo M (2014) Effects of proliferation and differentiation of mesenchymal stem cells on compressive mechanical behavior of collagen/β-TCP composite scaffold. J Mech Behav Biomed Mater 39:218–230
Cheng XG et al (2014) Platelet-derived growth-factor-releasing aligned collagen–nanoparticle fibers promote the proliferation and tenogenic differentiation of adipose-derived stem cells. Acta Biomater 10(3):1360–1369
Saxena N et al (2009) Patulin causes DNA damage leading to cell cycle arrest and apoptosis through modulation of Bax, p53 and p21/WAF1 proteins in skin of mice. Toxicol Appl Pharm 234(2):192–201
Zhao P et al (2013) Diethyl sulfate-induced cell cycle arrest and apoptosis in human bronchial epithelial 16HBE cells. Chem Biol Interact 205(2):81–89
Zhang J-Y et al (2013) Quercetin induces apoptosis via the mitochondrial pathway in KB and KBv200 cells. J Agric Food Chem 61(9):2188–2195
Acknowledgements
The author thanks the Ministry of Science and Technology (Taipei City, Taiwan, R.O.C.) for financial support (MOST-103-2621-M-244 -001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hsu, YC., Wu, CS., Liao, HT. et al. Improvement of the biocompatibility of polyhydroxyalkanoate by filling with hyaluronic acid. J Mater Sci 50, 7790–7799 (2015). https://doi.org/10.1007/s10853-015-9350-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-015-9350-0