
Abstract
ZnO nanoparticles are recently attracting a lot of interest due to their novel optical, electrical, mechanical, and chemical properties, due to surface and quantum confinement effects. A stable dispersion of ZnO nanoparticles with a uniform particle diameter of 5 nm has been synthesized via chemical precipitation. The surface of the particles has been functionalized with dodecanethiol for enhanced compatibility and dispersion in polymethylmethacrylate (PMMA). Transparent luminescent nanocomposite powders were obtained using solution mixing and solvent casting of ZnO nanoparticles in PMMA. This powder was used to produce injection-molded luminescent nanocomposites plaques. The effect of the ZnO nanoparticles on the polymer structure has been investigated by employing thermomechanical and dielectric spectroscopy analyses.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Zinc oxide (ZnO) is a member of the family of wide-gap semiconductors (room temperature band gap of 3.37 eV) and is ideal for short-wavelength optoelectronics applications [1]. ZnO typically exhibits emission bands in the ultraviolet (UV) and visible regions. The UV emission is usually considered as the characteristic emission of ZnO and is attributed to the band-edge transition. The emissions in the visible regions are universally considered to be associated with the intrinsic or extrinsic defects in ZnO [2].
Size reduction of ZnO into the nanoregime generates novel optical, electrical, mechanical, and chemical properties due to surface and quantum confinement effects [3, 4]. Recent applications of nanocrystalline ZnO in solar cells [5], field effect transistors [6], nanogenerators [7], and photocatalysis [8] make it a versatile material.
Polymethylmethacrylate (PMMA) is an optically clear amorphous thermoplastic. It is widely used as a substitute for inorganic glass, because it shows higher impact strength and undergoes ductile rather than brittle fracture [1, 9]; moreover, it has favorable industrial processing conditions and is one of the most scratch resistant commodity thermoplastics. PMMA transmits more light (up to 93 % of visible light) than glass and it does not filter ultraviolet light. It transmits UV light down to 300 nm and allows infrared light of up to 2800 nm to pass. PMMA has high mechanical strength, high Young’s modulus, and low elongation at break.
PMMA nanocomposites are widely studied not only because they show significant enhancements in mechanical properties, but also for the achievement of new multifunctional properties (e.g., optical, electrical, and thermal) that are not observed with micron-size fillers [9, 10].
In this paper, ZnO nanoparticles (NPs) have been synthesized by chemical precipitation at room temperature and the surface of the NPs has been functionalized with dodecanethiol for enhanced compatibility and dispersion in PMMA. In this way, the optical properties of the ZnO NPs could be retained through all processing steps. By performing a combination of structural characterization (transmission electron microscopy), thermomechanical, and dielectric characterization, it was possible to study the effect of the introduction of the ZnO NPs on the structure of the PMMA matrix and to elucidate the interaction between the PMMA matrix and the ZnO nanofillers.
Experimental
Materials
Zinc acetate dihydrate (Zn(CH3COO)2·2H2O),·4H2O), tetramethylammonium hydroxide (TMAH), 1-dodecanethiol, dimethyl sulfoxide (DMSO), toluene, and ethanol were supplied by Aldrich and used as received. PMMA was ALTUGLAS® VSE UVT (TP 727) supplied by Altuglas International.
Synthesis of NPs and nanocomposites
ZnO NPs were synthesized adapting the procedure presented by Guglieri and Chaboy [11]: 500 mg of Zinc acetate dihydrate (Zn(Ac)2) was dissolved in 22.5 mL3 dimethyl sulfoxide (DMSO) and 750 mg tetramethylammonium hydroxide (TMAH) was dissolved in 7.5 mL3 ethanol (EtOH). The TMAH solution was added dropwise (about 2 mL3/min) to the Zinc solution under vigorous stirring at room temperature; after 10 min, the solution was heated at 40 °C for 1 h to promote Ostwald ripening of the particles. Then the solution was cooled down to room temperature, 60 μL of dodecanethiol was then added and the precipitated NPs were then separated and collected by centrifugation (4000 rpm, 5 min), washed 2 times with acetone, and then redispersed in toluene.
The procedure employed for the preparation of the nanocomposite plaques is shown in Fig. S1 (see Supplementary material). First, the ZnO NPs were synthetized as previously described (step 1). Then the ZnO NPs dispersed in toluene (0.1 wt%) were added to a 100 mg/mL3 solution of PMMA in toluene and stirred for 12 h at room temperature. The mixture was then ultrasonicated for 30 min. (step 2). The combined solution was mixed vigorously for 1 min using a high-shear homogenizer and then immediately casted onto aluminum plates. The nanocomposites were dried at 60 °C overnight under nitrogen purge (step 3), obtaining nanocomposite powders incorporating 0.1 wt% of ZnO NPs. The luminescent plaques were produced by injection molding (step 4).
Plaques (60 × 10 × 0.95 mm) with 0.1 wt% ZnO NPs have been obtained. Plaques without ZnO NPs have also been produced. The sample without ZnO has been named sample I and the sample with 0.1 % ZnO sample II.
Characterizations
The crystalline phases of the ZnO NPs were characterized by X-Ray diffraction (XRD) using a Philips PW1710 diffractometer equipped with grazing incidence X-Ray optics. The analyses were performed at 0.5° incidence, using CuKα Ni-filtered radiation at 30 kV and 40 mA. The mean crystallite diameter has been evaluated using the Scherrer equation performing a Lorentzian fit of the most intense diffraction peaks.
Transmission electron microscopy (TEM) analysis of NPs deposited from colloidal solutions on carbon-coated copper grids was performed with a JEOL JEM 1010TEM operated at 100 keV. The nanocomposite samples were cut on a Leica Ultracut S ultramicrotone with a glass knife at room temperature and collected on 400 mesh copper grids.
UV–Vis absorption spectra of colloidal solutions were taken using a JASCO V-570 standard spectrometer. Total reflectance and transmittance spectra of the nanocomposite were recorded on a Jasco V-570 spectrophotometer equipped with a Jasco ISN-470 integrating sphere apparatus.
Photoluminescence (PL) emission (λ exc = 340 nm) spectra were measured by a Fluoromax-4 spectrofluorometer from Horiba Jobin–Yvon and were collected in the wavelength range of 300–900 nm.
The plaques were analyzed with Fourier transform infrared spectroscopy (FTIR), and spectra were taken in ATR configuration in the 700–4000 cm−1 region using a resolution of 2 cm−1.
A DSC2920 instrument (TA Instrument) equipped with an LNCA low temperature attachment was used for differential scanning calorimetry (DSC) measurements. Analyses were performed from −100 to +300 °C, with a 30 cm3 min−1 He flux and a heating rate of 10 °C min−1 on about 10 mg of sample hermetically sealed in an aluminum pan.
200-μm-thick films were prepared from sample I and II by hot-pressing for 5 min at 140 °C and 1000 psi. Dynamic mechanical and electrical responses were determined on the obtained thin films.
Dynamic mechanical analyses (DMA) were carried out with a TA Instruments DMA Q800 using the tension film clamp. Temperature spectra in the range from +20 to +130 °C at a rate of 4 °C min−1 were determined subjecting a rectangular film sample to an oscillatory sinusoidal tensile deformation at 1 Hz with amplitude of 4 μm. The mechanical response data were analyzed in terms of the elastic (storage) modulus (E’) and viscous (loss) modulus (E″). tanδ = E″/E′ was analyzed on temperature in order to reveal the mechanical relaxation phenomena.
Dielectric spectra were obtained by Alpha-A analyzer (Novocontrol) from 0.01 Hz to 10 MHz. Measurements were performed on the films sandwiched between two gold-plated electrodes, having diameter of 2 cm. Temperature was changed from 20 to 140 °C in 10 °C steps by a home-made thermostat operating with a liquid nitrogen flux.
Three point flexure tests were carried out on an Instron model 5543 universal testing machine using 3 replicates of each sample. Impact test measurements were performed with a Zwich/Roell pendulum. All the mechanical tests have been performed at 25 °C.
Results and discussion
Nanoparticles
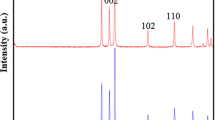
XRD pattern presented in Fig. 1 shows the presence of crystalline wurtzite hexagonal ZnO (JCPDS No. 36–1451). Crystallite mean diameter was calculated using the five values corresponding to the peaks recorded in the XRD pattern, and error was taken to be the standard deviation of these values. The estimated mean diameter is D = (6.3 ± 0.5) nm.

XRD pattern of ZnO NPs powder synthesized at 50 °C for 1 h
From TEM measurements (see Fig. 2), a mean particle diameter D = (6.0 ± 0.8) nm has been evaluated, in agreement with the size estimated by XRD .

TEM micrograph of ZnO NPs (top) and of PMMA-ZnO nanocomposite (bottom) showing an aggregate of ZnO NPs
Optical characterization of the colloidal solutions in DMSO shows a clear absorption onset in the near UV due to ZnO band gap and good transparency in the visible range as shown in Fig. 3. The PL spectra (Fig. 3) are dominated by the broad green emission associated with the intrinsic and extrinsic defects in ZnO [2].

Absorption (black line) and PL (gray line) spectra of ZnO NPs in DMSO (Color figure online)
Nanocomposite
TEM images displayed in Fig. 2 confirmed the presence of aggregates in the PMMA-ZnO nanocomposites with average size between 100 and 300 nm.
The optical quality of the nanocomposite plaques can be appreciated from Fig. 4, where the total transmittance spectra are reported. The absorption onset of pure PMMA appears at 250 nm, while that of the nanocomposite is broadened in the range of 350–390 nm, due to the absorption of the ZnO NPs [1, 12]. The total transmittance of the composite is in general very high. Pure PMMA has a transmittance of 93 % (@ = 600 nm). The loss of 7 % is caused by reflection on the two surfaces of the plaques (0.90 mm). PMMA with 0.1 % of ZnO has a transmittance of 91 % (@ = 600 nm).

Total transmittance of the sample I (PMMA; black line) and the sample II (PMMA-ZnO; gray line) (Color figure online)
Through all processing steps, the functionality of the ZnO NPs is fully retained, in fact the composites showed the typical green–yellow emission of ZnO NPs when irradiated with UV light (see Fig. 5).

Room temperature emission spectra of the sample I (PMMA; black line) and the sample II (PMMA-ZnO; gray line) (Color figure online)
FTIR spectra (see Fig. S2 in the supplementary material) showed the typical vibration bands of PMMA.
In Fig. 6, the DSC curves measured for sample I and II from −50 to 200 °C are shown. Two glass transitions, labeled as T g1 and T g2, were detected for each sample. In the case of sample I, T g1 and T g2 were localized at 66 and 93 °C, respectively, while for the sample II they were found at 42 and 64 °C, respectively (see Table 3). A wider change of the specific heat is associated to T g2 with respect to T g1. PMMA polymers usually do not crystallize, though they can show a certain degree of local order, as witnessed by the possibility to undergo a glass transition. The glass transition temperature can vary from 41 to 134 °C, depending on the polymer tacticity [13]. Molecular weight, nano-confinement, copolymerization, blending, and cross-linking can influence the glass transition temperature [14]. The presence of two different glass transition temperatures in both PMMA and PMMA-ZnO nanocomposite could be related to the existence of patchy micro-morphologies in the bulk of both materials, where domains composed of PMMA chains in different conformations could be distinguished. In the sample II, T g1 was detected at a relatively low temperature, typically corresponding to purely iso-tactic PMMA domains (see A domains in Fig. 7). T g1 of sample I and T g2 of sample II were localized at temperatures ranging from 60 to 70 °C, thus suggesting the presence of domains in which more than 50 % of the PMMA polymer chains assume the iso-tactic conformation (see B domains in Fig. 7).

DSC profiles for sample I (PMMA; top black line) and sample II (PMMA-ZnO; bottom gray line). The straight lines were added as guide to the eye (Color figure online)

Morphological model proposed for sample I (PMMA) and sample II (PMMA-ZnO). A iso-tactic PMMA domains; B domains of PMMA with 50 % of iso-tactic conformation; C PMMA domains with <25 % iso-tactic, ca. 40 % syndio-tactic, ca. 35 % hetero-tactic conformation; D ZnO domains
The semi-quantitative evaluation of PMMA conformation was performed using the data reported in Ref. [12] (at page V/88) where the glass transition temperature of PMMA with 62 % of iso-tactic conformations, 20 % hetero-, and 18 % syndio-tactic is at 61.6 °C. By comparison with this datum, it was possible to evaluate that the domains in sample II responsible of the T g detected between 60 and 70 °C should be characterized by a conformational composition similar to that reported in the literature, thus those domains contain probably iso-tactic chains in a concentration of more than 50 %.
The localization of T g2 of sample I at 93 °C suggested the presence of domains in which the iso-tactic PMMA chain conformation is less than 25 %. More than 40 % of the chains are in syndio-tactic chain conformations and the remaining chains are in hetero-tactics (see C domains in Fig. 7). It can be concluded that with respect to sample I, the morphology of sample II includes a higher fraction of chains in iso-tactic conformation. It can be hypothesized that the inorganic filler in sample II triggers the formation of iso-tactic conformation in PMMA polymer chains which give a wider interaction between the strongly polar lateral chains of the PMMA and the inorganic filler.
Results of DMA are shown in Fig. 8, in terms of storage modulus (E′) and loss modulus (E″). The storage modulus decreases as the temperature increases for both materials. For temperatures ranging from 20 to 50 °C, E’ is slightly higher for the sample II with respect to the sample I. It is reasonable to assume that in macroscopic samples, E′ can be correlated to both the chain conformation geometries composing PMMA domains which are characterized by the weakest inter-chain bonding interactions and by the inter-domain interactions. In the sample II, the inorganic–organic, dipole–dipole, or ion–dipole interactions are responsible for the improved inter-domain interactions with respect to sample I. Therefore, the weaker mechanical response of sample I with respect to sample II should be reasonably related to the inter-domains interactions which are stronger and more abundant in the sample II.

DMA graphs of sample I (PMMA—black line) and sample II (PMMA-ZnO—gray line) (Color figure online)
For both materials, E′ shows a steep decrease, localized at 90 °C in the sample I and at 76 °C in the sample II. Correspondingly, a peak is revealed in the E″ graph, for both materials (see Table 1). This effect can be attributed to the second glass transition T g2 and after the glass transition completion both materials show reduced mechanical strength due to the glassy states.
Results of the dielectric analysis are shown in Fig. S3 (see Supplementary material), in terms of the real component of the permittivity (ε′) (top) and tanδ (bottom) versus temperature for some selected frequencies. ε′ values are in the range of 2.5–7 and increase with increasing temperature and with decreasing frequencies, in accordance with other measurements performed on methacrylate-based polymers [13, 14]. The dependence of ε′ versus temperature shows a step-shaped profile (Fig. S3). The step-like increase of ε′ is coincident with the glass transition temperature T g2.
For temperatures from 20 to 60 °C ca., ε′ values were slightly higher for sample I with respect to sample II. This fact can be ascribed to the distribution of conformational states which characterize the micro-morphology of the sample II. Indeed in sample II, the higher extent of iso-tactic polymer chains increases the density of PMMA-filler interactions reducing the density of free dipoles of side groups present in sample I.
For temperatures higher than 60 °C, ε′ is higher in sample II than in sample I. Therefore, the glass transition in sample II starts at lower temperatures with respect to sample I. For temperatures higher than 120 °C, indeed, the glass transition is completed for both the materials and ε′ is again higher for sample I.
In heterogeneous materials, inter-domain polarization occurs as a result of the accumulation of virtual charges at each interface between nano-domains that have different dielectric constants. The accumulation of free charges is responsible for the field distortion and the dielectric loss of energy, which causes a significant increase in ε′ [15].
The bottom of Fig. S3 (see Supplementary material) shows the tanδ (ε″/ε′) profiles versus temperature for both samples. Two peaks can be easily distinguished in the spectra measured at 100 Hz. The peaks are assigned to the α and β relaxations, respectively, at low and high temperature [16]. α is assigned to the segmental motion of polymer backbone chains and β to the relaxation of PMMA side chains.
It can be observed that the values of tanδ are practically the same for both materials, except for profiles at 1 Hz and for temperatures higher than ca. 80 °C. In these conditions, tanδ values of sample II are clearly higher than that of sample I, of about one order of magnitude. This phenomenon is associated to the polarization effects generated at the interfaces between PMMA and ZnO NPs, which probably makes Zn2+ cations mobile along the PMMA domains.
The imaginary component of the electrical permittivity of the two materials is shown in Fig. 9 as a function of frequency. The most evident feature in the spectra is the presence at f > 100 Hz of an intense relaxation peak labeled β 1, which shifts at higher frequencies as the temperature is increased. The α relaxation is detected on the low frequencies side of the β 1 peak.

Imaginary component of the electrical permittivity of sample I (PMMA) and sample II (PMMA-ZnO) as a function of frequency for increasing temperatures from 20 °C up to 140 °C
In addition, a further peak is observed in the high-frequency side of the β 1 peak which is labeled as β 2 (see Fig. 9). At T > 90 °C for sample II and at T > 100 °C for sample I, a significant conductivity contribution is detected.
The most intense relaxation observed in the PMMA-based materials by means of both dielectric and dynamic mechanical measurements is often considered as the archetype of a localized (β) relaxation in polymers [17]. This β relaxation provides a mechanism for energy dissipation which can be linked to PMMA’s mechanical properties from room temperature up to the T g.
β peak is assigned to hindered rotational fluctuation relaxations around the bond linking the side group to the main chain. Restricted uniaxial rotation events of polymer backbone coupled to the 180° reorientational fluctuation mode of the side-chains assigned to the β-process have been observed at the glass transition temperature. The large dipole moment of the carboxyl side group justifies the significant relaxation intensity exhibited by this peak [17]. It is generally recognized that this relaxation is strongly related to the degree of polymerization, tacticity, side-chain length, nano-confinement, and cross-linking phenomena in bulk polymer [18].
The presence in both samples of the β 2 relaxation is related to the population of side chain present in some inter-domain regions, where a higher free volume can be hypothesized and consequently faster fluctuation flip motions with respect to the bulk conventional PMMA are expected.
The relaxation phenomena above assigned are studied in greater details by fitting their dielectric spectra profiles with the following general equation [19]:
where σ 0 is the dc conductivity, ε 0 is the vacuum permittivity, ω (ω = 2πf and f is the frequency in Hz) is the angular frequency of the electromagnetic field, Δε k is the dielectric strength of the k-th dielectric relaxation, τ k is the characteristic time of the k-th dielectric relaxation, m k is the shape parameter of the k-th dielectric relaxation, and ε ∝ is the permittivity limit at high frequency.
The Nyquist plot of the electric response for material II, in terms of permittivity, at 30 and 50 °C is reported in Fig. S4 (see Supplementary material) from which it is possible to appreciate the fitting corresponding to the equivalent circuit described by Eq. (1).
The dependence of σ 0 on the inverse of the temperature is shown in Fig. 10a. The σ 0 values for sample II are higher than that of sample I. These results suggest that the presence of ZnO NPs increases the density of mobile charge species. Traces of dissociated ions generated by small amounts of ZnO in PMMA-ZnO nanocomposite can be responsible for the higher conductivity of the nanocomposite with respect to the pure PMMA.

a d.c. conductivity determined by fitting of the dielectric spectra of sample I (PMMA) and sample II (PMMA-ZnO) with Eq. (1). b relaxation times of the sample I (PMMA) and sample II (PMMA-ZnO)
The temperature dependence of σ 0 is satisfactorily reproduced by a Vogel-Tamman-Fulcher (VTF) [20] equation, by means of the parameters reported in Table 2. It should be highlighted that the pseudo-activation energy determined from the conductivity data of sample II is about half than that of pristine polymer. The presence of the filler seems to facilitate the free charge migration process. As mentioned above, the filler is mostly expected to be localized in the inter-domain regions. The size of these inter-domain regions seems to be bigger in sample II than in sample I. It should also be noted that the E a and the T 0 values reported on Table 2 confirm the above-proposed structural hypotheses.
The characteristic relaxation times of α and β modes obtained from Eq. (1) are reported in Fig. 10b with respect to the inverse of the temperature. It should be noted that the τ k relaxations are of the same order of magnitude for both sample I and II and that the major differences are detected for the α relaxation events. The α relaxation mode was faster in sample II than in sample I. This result is in accordance with the above-mentioned structural hypotheses, which suggests higher iso-tactic polymer chain content in sample II with respect to sample I. As reported elsewhere [21], the polymer chains in the iso-tactic conformation are characterized by weaker inter-chain interactions than the syndio- or hetero-tactic chain conformations. In detail, syndio-tactic PMMA is more entangled, possesses lower chain mobility and a smaller coil size.
The temperature dependence of the α relaxation times is satisfactorily fitted by a Vogel-Tamman-Fulcher-Hesse (VTFH)-type equation. The obtained pseudo-activation energies are reported in Table 2. It can be observed that they are nearly coincident for both materials (about 10 kJ/mol). The value of the ideal glass transition temperature is higher for sample I than that for sample II. In polymers, T 0 is usually revealed at 50 °K below T g [21]. Thus, as expected, if we add 50 °K to the T 0 of sample I reported in Table 2 a value of T g2 close to that measured by DSC is obtained, which confirms that the observed α relaxation is correlated to the main glass transition of the material.
For sample II, the pseudo-activation energy and the T 0 values determined from the conductivity profile are practically coincident with the values determined from the α relaxation time profile. This result suggests that the ionic species migration in sample II is assisted by the segmental motion of the PMMA backbone chains.
The β relaxation rate is not affected by the inorganic filler in the composite material. The dependence on temperature of β relaxation time shows an Arrhenius-type behavior, with coincident activation energies for both materials.
Two temperature regions are distinguished for the β relaxation. In the high-temperature region, the value of the activation energy is twice that determined for the low-temperature region. The two temperature regions are delimited at 100 °C for sample II and at 120 °C for sample I. This difference is explained if we consider that the relaxation motion of polymer side chains is stimulated thermally in different ways. In detail, the main backbone chains present a higher mobility in sample II, a glass transition temperature at lower temperatures and a relatively higher content of backbone chains in iso-tactic conformation.
The dependence on the inverse of the temperature of dielectric strength is shown in Fig. 11a. It should be noted that the Δε trends are similar for both the investigated materials. The intensity of the β 1 relaxations increases as the temperature increases, for both the materials, while for the α relaxation it decreases as the temperature increases. The intensity of β 2 relaxation is weaker than that of β 1.

a Dielectric strength of the relaxation phenomena and b shape parameter of the relaxations events of sample I (PMMA) and sample II (PMMA-ZnO)
This result indicates that as the temperature increases, the density of the interactions of side groups with the environment decreases, while for the α relaxation the opposite behavior is observed.
The dependence of the shape parameters m k on the inverse of the temperature is shown in Fig. 11b for the α e β 1 relaxations. The β 2 relaxation shape parameter was fixed at 0.34. Figure 11b shows that the shape parameters for the β relaxations, are ranging from 0.3 to 0.6, as expected for typical polymeric materials. The shape parameter of β 1 increases with increasing temperatures, i.e., the band shrinks revealing that the distribution of relaxation time becomes more and more narrow. Thereby β 1 is associated with a dipole moment which, as the temperature rises, reduces its interactions with the neighbor environment. The shape parameters of the β 1 relaxation are higher for sample II with respect to sample I, which confirm that the mobility of functional groups of sample II is higher than that of sample I. This evidence is easily understood if we admit that smaller domains are present as a result of the interaction between the side groups and the ZnO nanofiller.
A summary of the three point flexure test and impact test of the nanocomposites is provided in Table 3 from which it is possible to note that the PMMA-ZnO nanocomposite is more rigid than the pure PMMA, in accordance with the proposed model.
Conclusions
ZnO NPs have successfully dispersed in PMMA and their optical properties have been transferred to the PMMA-ZnO nanocomposites.
The influences of PMMA–PMMA and PMMA-ZnO interactions on the macroscopic properties of pure PMMA and PMMA-ZnO nanocomposite have been studied by thermal, mechanical, and electrical characterization methods.
The DSC profiles show two glass transitions temperatures (T g1 and T g2). In the PMMA-ZnO nanocomposite, the glass transition temperatures were detected at about 25 °C temperatures lower than that of pure PMMA.
The results are explained on the basis of the interactions and structural features of the materials. Two different domains are distinguishable in both materials which are characterized by a different conformational distribution of the PMMA chains. Indeed, the iso-tactic component resulted to be higher in the nanocomposite than in the pure PMMA. The inorganic nanofiller, which is localized mostly at the interfaces between nano-domains, interacts with the polar side chains of the polymer matrix thus increasing the concentration of iso-tactic conformation of backbone chains.
The thermos-mechanical and dielectric measurements showed that the α relaxation process, which is associated to the segmental motion of the polymer backbone chains, is correlated to the main glass transition temperature and, in the case of the PMMA-ZnO nanocomposite, it is clearly responsible for the long-range-conductivity of mobile species. The typical β relaxation of PMMA is revealed for both samples, by the mechanical and the electrical characterization. Nevertheless, only in the electrical profile response, it was possible to see the presence of a further very week β relaxation. The detailed analysis of the β modes dependences on temperature confirmed that the amount of iso-tactic conformation is higher in the PMMA-ZnO sample with respect to the pure PMMA sample.
The reported results demonstrate that the interactions between PMMA and ZnO NPs trigger the increase of the fraction of iso-tactic conformation polymer chains in PMMA matrix which is responsible of the higher mobility and the better mechanical and the thermal properties (lower T g values) of the nanocomposite.
References
Sun D, Miyatake N, Sue HJ (2007) Transparent PMMA/ZnO nanocomposite films based on colloidal ZnO quantum dots. Nanotechnology 18:15606
Zeng H, Duan G, Li Y, Yang S, Xu X, Cai W (2010) Blue luminescence of ZnO nanoparticles based on non-equilibrium processes: defect origins and emission controls. Adv Funct Mater 20:561–572
Wang ZL (2004) Zinc oxide nanostructures: growth, properties and applications. J Phys 16:R829–R858
Djurisic AB, Leung YH (2006) Optical properties of ZnO nanostructures. Small 2:944–961
Law M, Greene LE, Johnson JC, Saykally R, Yang P (2005) Nanowire dye-sensitized solar cells. Nat Mater 4:455–459
Suh DI, Lee SY, Hyung JH, Kim TH, Lee SK (2008) Multiple ZnO nanowires field- effect transistors. J Phys Chem C 112:12761281
Wang XD, Song JH, Liu J, Wang ZL (2007) Direct-current nanogenerator driven by ultrasonic waves. Science 316:102–105
Barreca D, Ferrucci AP, Gasparotto A, Maccato C, Maragno C, Tondello E (2007) Temperature-controlled synthesis and photocatalytic performance of ZnO nanoplatelets. Chem Vap Depos 13:618–625
Demir MM, Memesa M, Castignolles P, Wegner G (2006) PMMA/Zinc oxide nanocomposites prepared by in situ bulk polymerization. Macromol Rapid Commun 27:763–770
Thakur VK, Thunga M, Madbouly SA, Kessler MK (2014) PMMA-g-SOY as a sustainable novel dielectric material. RSC Adv 4:6677–6684
Guglieri C, Chaboy J (2010) Characterization of the ZnO–ZnS interface in THIOL-capped ZnO nanoparticles exhibiting anomalous magnetic properties. J Phys Chem C 114:19629–19634
Li S, Toprak MS, Jo YS, Dobson J, Kim DK, Muhammed M (2007) Bulk synthesis of transparent and homogeneous polymeric hybrid materials with ZnO quantum dots and PMMA. Adv Mater 19:4347–4352
Brandrup J, Immergut EH, Grulke EA, Abe A, Bloch DR (1999) Polymer handbook, 4th edn. Wiley, New York
Ngai KL, Gopalakrishnan TR, Beiner M (2006) Relaxation in poly(alkyl methacrylate)s: change of intermolecular coupling with molecular structure, tacticity, molecular weight, copolymerization, crosslinking, and nanoconfinement. Polymer 47:7222–7230
Di Noto V, Giffin GA, Vezzù K, Piga M, Lavina S (2012) Broadband dielectric spectroscopy: a powerful tool for the determination of charge transfer mechanisms in ion conductors. In: Knauth P, Di Vona ML (Eds) Solid state proton conductors: properties and applications in fuel cells. Wiley, Weinheim, pp. 107–180
Gross S, Camozzo D, Di Noto V, Armelao L, Tondello E (2007) PMMA: a key macromolecular component for dielectric low-κ hybrid inorganic–organic polymer films. Eur Polym J 43:673–696
Schmidt-Rohr K, Kulik AS, Beckham HW, Ohlemacher A, Pawelzik U, Boeffel C, Spiess HW (1994) Molecular nature of the beta relaxation in poly(methyl methacrylate) investigated by multidimensional NMR. Macromolecules 27:4733–4745
Wind M, Graf R, Renker S, Spiess HW (2005) Structural reasons for restricted backbone motion in poly(n-alkyl methacrylates): degree of polymerization, tacticity and side-chain length. Macromol Chem Phys 206:142–156
Barsoukov E, Macdonald JR (2005) impedance spectroscopy theory, experiment, and applications, 2nd edn. Wiley, New York
Di Noto V, Barreca D, Furlan C, Armelao L (2000) Zeolitic inorganic-organic polymer electrolytes: a material based on poly(ethylene glycol) 600, SnCl4 and K4Fe(CN)6. Polym Adv Technol 11:108–121
Fuchs K, Friedrich Chr, Weese J (1996) Viscoelastic properties of narrow-distribution poly(methyl methacrylates). Macromolecules 29:5893–5901
Acknowledgements
AM wishes to thank EU for funding through the 7FP EU Project—ORION #222517.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dai Prè, M., Martucci, A., Martin, D.J. et al. Structural features, properties, and relaxations of PMMA-ZnO nanocomposite. J Mater Sci 50, 2218–2228 (2015). https://doi.org/10.1007/s10853-014-8784-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8784-0