
Abstract
The magnetism and electronic transport properties of \(\hbox {La}_{0.6}\hbox {A}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (A = Sm, Dy, Er) are studied by the measurements of magnetization and resistivity. Infrared spectra reveal that two typical absorbed peaks occur in their corresponding positions. However, the stretching mode \(\nu _{3}\) exhibits an obvious shift to low frequency and its intensity gets enhanced in Dy- and Er-doping samples. In Raman spectra, the stretching mode shows the same shift, while the Jahn–Teller mode remains invariant. The A-site substitution with other elements changes the average ionic radius and induces cation disorder, causing a decrease of the Curie temperature and the formation of spin–cluster-glass. Except for Sm-doping sample, the field dependence of magnetization at different temperatures shows a similar magnetization process indicating that the low concentration substitution with 10 % magnetic ions cannot signally influence the magnetic exchange interaction on B-site sublattice. Based on the analysis of electronic transport in metallic regime, we find that the main factor for impacting the electronic transport is from the variation of crystal lattice and the induced cationic disorder, regardless of magnetic or non-magnetic substitution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past two decades, mixed-valence perovskite manganites \(\hbox {A}_{1 - x}\hbox {B}_{x}\hbox {MnO}_{3}\) (A = rare earth element, B = divalent alkaline earth element) have attracted tremendous interest due to their unique physical properties, stemming from the close interplay of spin, charge, orbital, and lattice degree of freedom [1–5]. Extensive efforts to understand these mechanism have shown that the doping concentration x controls the band filling and the \( {e}_{\rm g}\) electron transfer. The stoichiometric \(\hbox {LaMnO}_{3}\) is an antiferromagnetic insulator with A-type antiferromagnetism. By partial substitution with divalent elements B for La, a corresponding amount of \(\hbox {Mn}^{3+}\) was converted into \(\hbox {Mn}^{4+}\), leading to the appearance of mixed-valence state \(\hbox {Mn}^{3+}/\hbox {Mn}^{4+}\). Under proper condition, the double-exchange (DE) interaction between \(\hbox {Mn}^{3+}\) and \(\hbox {Mn}^{4+}\) can cause a strong paramagnetic–ferromagnetic (PM–FM) transition together with an insulator–metal (IM) transition around the Curie temperature. In this regime, the well-known colossal magnetoresistance has been frequently observed [6–8]. Meanwhile, a viewpoint of the electronic/magnetic phase separation due to the intrinsically inhomogeneous and quenched disorder was proposed to explain the PM–FM phase transition together with IM transition in perovskite manganites [9]
Although the magnetism and electronic conductivity of manganites are sensitive to the \(\hbox {Mn}^{3+}/\hbox {Mn}^{4+}\) ratio, some pioneer investigations have indicated that they are also affected strongly by A-site average cationic radius \(\langle {r}_{\rm A}\rangle \) and cationic disorder which is quantified by the size variance of the A-site cation radius distribution \(\sigma ^{2}\). (\(\langle {r}_{\rm A}\rangle =\sum {x}_{\rm i} {r}_{\rm i}, \,\,\,\sigma ^{2}=\sum {x}_{\rm i} {r}_{\rm i}^{2}-\langle {r}_{\rm A}\rangle ^{2},\) where \({x}_{\rm i}\) is the fractional occupancy of A-site ions and \({r}_{\rm i}\) is the corresponding ionic radius [10, 11]. Generally, with the increase in average cationic radius \(\langle {r}_{\rm A}\rangle \), the Mn–O–Mn bond angle will be enlarged, giving rise to an enhancement of one-electron bandwidth and the Curie temperature \({T}_{\rm C}\). On the other hand, A-site cationic disorder \(\sigma ^{2}\) is another important factor for influencing the magnetism and electronic transport properties of manganites [12, 13]. Due to the existence of cationic disorder, A-site ionic substitutions change the pristine DE interaction or induce a new magnetic interaction between A- and B-sublattice. However, many previous works have mainly focussed on the understanding of the different A-site cationic radii and disorder effects which influence magnetic and electronic transport properties in manganites. On the contrary, the A-site doping elements with or without magnetism have not been paid much attention to. In fact, the A-site ionic magnetism is closely related to the systemic magnetic–electronic properties. Generally, as some larger magnetic ions occur on A-site, they more or less change and influence \(\hbox {Mn}^{3+}{-}\hbox {O}^{2-}{-}\hbox {Mn}^{4+}\) exchange interaction on B-site. Moreover, this influence even exceeds the other factors including A-site average cationic radius and cationic disorder. A-site substitution with magnetic ion causes a decrease in the Curie temperature and the weakening of ferromagnetism [14–17]. What is more, other novel behaviors have been observed. Sun et al. and Guo et al. reported that the spin-canted structure and ferrimagnetic state occurred in the Gd-doped \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) and \(\hbox {La}_{0.67}\hbox {Ca}_{0.33}\hbox {MnO}_{3}\) [18, 19]. A possible quantum critical point in the Dy-doping \(\hbox {La}_{0.7}\hbox {Ca}_{0.3}\hbox {MnO}_{3}\) was reported by Yusuf et al. [20]. Recently, a tricritical point has been found in the \(\hbox {La}_{0.7-x}\hbox {Nd}_{x}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (x = 0.6) [21]. Therefore, the A-site substitutions with magnetic ion possibly play a more important role in influencing the systemical properties. Hence, in this paper, we chose three different magnetic elements of Sm, Dy, and Er to replace La with 10 % concentration. Their magnetic moments are 10.6 (\(\hbox {Dy}^{3+}\)), 9.5 (\(\hbox {Er}^{3+}\)), and 1.5 \(\mu _{\rm B}\) (\(\hbox {Sm}^{3+}\)). Depending on their different magnetic moments and different cationic radii, we can further clarify which is the main effect on the magnetic and electron transport properties. From the measurements of infrared and Raman spectra, we find that the stretching mode \(\nu _{3}\) shows an obvious shift against the bending mode \(\nu _{4}\) in Dy- and Er-doping samples. These results indicate that there are large \(\hbox {MnO}_{6}\) octahedron distortion and strong electron-phonon interaction in Dy- and Er-doping samples. In the temperature-dependent magnetization curves, except for the lower Curie temperature \({T}_{\rm c}\), all samples show a similar PM–FM phase transition, namely the magnetization displays a significant increase at their respective Curie temperatures. Moreover, for the substitution with large magnetic moment of Dy- and Er-doping samples, their isothermal magnetization at high and low temperatures all show a rapid increment and reach saturation at \(H\ge 0.5\) T, indicating A-site cationic magnetism is not enough to signally influence and change the whole systemic magnetic behavior. Similarly, from the analysis of electronic transport, we can find that the metal-like conductivity only reflects the effect of electron–electron and electron–phonon interactions rather than an additional magnetic scattering. We suggest that, in A-site magnetic doping with low concentration, the lattice effect should play a main role in deciding the magnetic and electronic properties.
Experiment
Polycrystalline samples \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO) were prepared by the conventional solid-state reaction method [22]. The mixture was preheated in air at 900 °C for 24 h. Afterward, the powder was ground and heated at 1200 °C for 30 h. Finally, it was reground, pressed into pellets, and sintered for another 40 h at 1400 °C, and was cooled down to room temperature with the furnace. The structure and phase purity of as-prepared samples were checked by powder X-ray diffraction (XRD) using Cu Kα radiation at room temperature. The measurements of infrared (IR) transmission spectra were carried out at room temperature with powder samples in which KBr was used as a carrier. Raman scattering spectra were obtained on a Spex Ramanlog 6 Raman spectrophotometer using a back-scattering technique. Temperature dependence of magnetization (M) was measured using a superconducting quantum interference device under 100 Oe magnetic field. The resistance \(\rho \) was measured by standard four-probe method.
Results and discussion
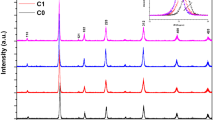
The structure and phase purity of the samples were examined by XRD measurements. As shown in Fig. 1, the XRD patterns reveal that all samples are in single phase with rhombohedral structure. Obviously, the doping ions have partly replaced \(\hbox {La}^{3+}\) ions and occupied A-site in the \(\hbox {ABO}_{3}\) structure. Figure 2 displays the Rietveld refinement of LSMO powder XRD pattern which can confirm that the sample is in single phase with a rhombohedral structure (\( {R}\bar{3} {c}\) space group). In order to investigate the effect of A-site average cationic radius \(\langle \hbox {r}_{A}\rangle \) and cationic disorder \(\sigma ^{2}\) on magnetism and electronic transport properties in all doping samples, these two parameters are calculated and listed in Table 1. The weighted average cationic radius \(\langle {r}_{\rm A}\rangle \) and A-site cationic disorder \(\sigma ^{2}\) were calculated using nine-coordinated ionic radii given by Shannon [23]. As we know, A-site average cationic radius \(\langle {r}_{\rm A}\rangle \) decides the variation of Mn–O–Mn bond angles and Mn–O bond lengths. Generally, the small \(\langle {r}_{\rm A}\rangle \) decreases the Mn–O–Mn bond angles and increases the Mn–O bond lengths [24], which weakens the hopping integral of \( {e}_{\rm g}\) electrons and attenuates the DE interaction between \(\hbox {Mn}^{3+}/\hbox {Mn}^{4+}\). As a result, the electronic bandwidth W gets narrow. From Table 1, one can find that the average cationic radius \(\langle {r}_{\rm A}\rangle \) slightly decreases from \(1.2442\, {\AA }\) for LSMO to \(1.2288\, {\AA }\) for LSEMO. A-site cation disorder \(\sigma ^{2}\), which mainly causes the random displacements of oxygen atoms from their average crystallographic positions [25], exhibits an increase from LSMO to LSEMO. IR spectra are very sensitive to local lattice distortions. For LSMO with \(\hbox {ABO}_{3}\) structure, Mn and its nearest-neighbor six \(\hbox {O}^{2-}\) form \(\hbox {MnO}_{6}\) octahedron with \(\hbox {O}_{h}^{6}\) symmetry, which has six vibrating modes [26]. Meanwhile, only the stretching mode \(\nu _{3}\) and the bending mode \(\nu _{4}\) are IR activated. Their vibrating frequencies are about \(\nu _{4}\sim 350\) and \(\nu _{3}\sim 600\; \hbox {cm}^{-1}\), respectively. Figure 3 depicts the observed IR spectra for all samples. In the present detecting frequency range, two absorbed peaks of \(\nu _{3}\) and \(\nu _{4}\) occur in their corresponding positions. The stretching mode \(\nu _{3}\) is related to the Mn–O bond lengths, while the bending mode \(\nu _{4}\) is related to the Mn–O bond angles. In Fig. 3, the \(\nu _{4}\) shows a little blue-shift for Dy- and Er-doping samples implying a slight variation of Mn–O–Mn bond angles. However, the \(\nu _{3}\) exhibits a more visible absorption peak for Dy- and Er-doping samples than for Sm-doping sample. Moreover, their absorption peaks also exhibit a slight shift to low-frequency region, indicating that the stretching mode becomes more prominent with the increase of \(\sigma ^{2}\). It also implies that the substitution with Dy and Er mainly causes a change of Mn–O bond lengths instead of Mn–O–Mn bond angles. Therefore, the variation of Mn–O–Mn bond angles can be ignored. The variation of Mn–O–Mn bond angles is decided by the distorted degree of \(\hbox {MnO}_{6}\) octahedron which is related to Jahn–Teller (JT) effect (tetragonal distortion). It can be inferred that the substitutions with Sm, Dy, and Er for 10 % La do not influence remarkably the \(\hbox {MnO}_{6}\) octahedron of LSMO. Furthermore, it is conceivable that if we do not consider other factors, the metallic-like electronic transport in three doping samples should show a similar conductivity since the Mn–O–Mn bond angles decide the \( {e}_{\rm g}\) electron transfer integral.

X-ray diffraction patterns of \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3},\; \hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3},\; \hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (Color figure online)

The Rietveld refinement of powder XRD patterns of \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) using GSAS, with \( {R}_{\rm p}=5.414\, \%\) and \( {R}_{\rm wp}=8.532\, \%\). The vertical marks indicate the position of Bragg peaks and the bottom curves show the difference between the observed and calculated intensities (Color figure online)

Infrared phonon spectrum (\(\nu \) represents infrared spectra frequency) of \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO) (Color figure online)
In order to testify the above inference, we performed the measurements of Raman spectra on all samples. As shown in Fig. 4, two Raman peaks appear around 480 and \(650 \,\,\hbox {cm}^{-1}\), which correspond to the \( {A}_{\rm g}\) (JT Mode) and \( {B}_{\rm 2g}\) (breathing mode), respectively [27, 28]. The former is out-of-phase bending, while the latter is in-phase stretching. The JT modes of four samples are almost at \(480 \,\hbox {cm}^{-1}\) and their positions do not change, indicating that the JT distortion of \(\hbox {MnO}_{6}\) octahedron has no much difference in all samples. However, the \( {B}_{\rm 2g}\) peak of stretching mode shows a perceptible shift to lower wave number. Moreover, this shift is particularly evident in Dy- and Er-doping samples, consistent with the strong absorbed peak and the same shifts to low wave number in their IR spectra.

Raman spectrum of \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO) (Color figure online)
Figure 5a shows the temperature dependence of magnetization (M–T) of LSMO measured under the magnetic field of 100 Oe. All data were measured on the warming process after zero-field cooling (ZFC, red circles) and field cooling (FC, blue circles), respectively. The M–T curve exhibits a sharp PM–FM phase transition, which is consistent with that in-phase diagram reported by Pollert et al. [29] The Curie temperature (\( {T}_{\rm C}\)), defined by the minimum in dM/dT, has been determined to be 368 K (see the inset of Fig. 5a). Figure 5b–d present the M–T curves of the doping samples. All samples exhibit a rigid PM–FM phase transition and their Curie temperatures have also been deduced from the corresponding M–T curves. As shown in insets of Fig. 5, the Curie temperatures of doping samples are driven to low temperature, which is consistent with the previous studies on the same A-site substitution with Sm, Dy, and Er [14–17]. As we know, the smaller average ionic radius brings the larger lattice distortion. The usual interpretation is that the smaller average ionic radius causes a larger distortion of the Mn–O–Mn bond lengths or angles. Thus, the DE interaction is weakened and the electron bandwidth gets narrow. Here, the average cationic radius on A-site (\(\langle {r}_{\rm A}\rangle \)) decreases from LSSMO to LSEMO. It is natural to find that their Curie temperatures decrease from 340 to 300 K. In Fig. 5, one can also find that the FC curve does not overlap ZFC curve at \( {T}<{T}_{\rm C}\) and exhibits an obvious irreversibility for all doping samples. Generally, the large difference between ZFC and FC magnetization is usually understood as a sign of spin–cluster-glass state that has no simple long-range ferromagnetic ordering. The main reason can be ascribed to large magnetic anisotropy and magnetic order frustration. In the current doping samples, the \(\hbox {Sm}^{3+}\), \(\hbox {Dy}^{3+}\), and \(\hbox {Er}^{3+}\) cations form a random distribution on A-site sublattice. It inevitably generates some magnetic domains with different sizes and forms some magnetic inhomogeneity in the system. In the solid-state synthesis, the ionic random distribution is a common phenomenon. For example, in LSSMO sample, the area with rich \({\rm La}^{3+}\) ions can form more \({\rm La}_{0.7}{\rm Sr}_{0.3}{\rm MnO}_{3},\)whereas the area with rich \({\rm Sm}^{3+}\) ions can form more \({\rm Sm}_{0.7}{\rm Sr}_{0.3}{\rm MnO}_{3}\). Both of them coexist in the same material. They have different magnetic structure and magnetic phase transition temperature. Therefore, their mutual competition hinders the magnetic domain motion. As a result, the spin–cluster-glass state occurs in the low-temperature region. Moreover, the A-site substitution with magnetic ion can directly influence the B-site magnetic exchange interaction due to coupling between two sublattices. Here, both \(\hbox {Dy}^{3+}\) and \(\hbox {Er}^{3+}\) ions have a large local moment of 10.6 and 9.5 \(\mu _{\rm B}\), respectively. In order to clarify this issue, we performed the measurements of isothermal magnetization at two different temperatures. One is at 5.0 K and the other is at 200 K, slightly below \(T_{\rm c}\). As shown in Fig. 6c, d, their magnetizations all exhibit a rapid increment and become saturated at \(H>0.5\) T, but there are no any magnetic hysteresis to be observed. Although both M(H) curves show a similar variation, their saturation magnetizations at two temperatures reveal a distinct difference and the saturation magnetization at 5 K is larger \({\sim}10\) emu/g than that at 200 K. Furthermore, for LSSMO sample in Fig. 6b, its magnetization is very small and does not reach saturation under high magnetic field, indicating that the low concentration of \({\rm Sm}^{3+}\) ions produce more stronger effect to change the DE interaction on B-site sublattice. Therefore, the B-site FM exchange interaction has been signally influenced by the A-site doping. However, from the comparative analysis of Fig. 6b–d, one can find that the doping magnetic ions should not play a main role in influencing the systemical magnetic coupling. We consider that the possible reason is due to the low doping concentration (10 %) and the random distribution of magnetic ion on A-site, which makes the doping magnetic ions unable to form an effective FM coupling to change the B-site FM interaction. Therefore, the induced disorder effect should be an important reason for suppressing the the DE interaction on B-site sublattice. In fact, the magnetization decrease due to disorder effect is not confined to the A-sited doping, which have been also frequently observed in the B-sited doping system [30, 31].

Temperature T (K) dependence of magnetization M (emu/g) measured under 100 Oe magnetic field for \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO). Red symbols are for zero field cooling and blue symbols are for field cooling. The inset is first derivative of magnetization (Color figure online)

Magnetic field H (T) dependence of magnetization M (emu/g) at 200.0 and 5.0 K for \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO) (Color figure online)
Figure 7 presents the temperature dependence of resistivity \(\rho \) for all samples. A small peak occurs in the \(\rho {-}T\) curves indicating that these samples undergo an IM transition. Their IM transition temperatures are 346, 318, and 325 K, respectively. At \(T>T_{\rm IM}\), the temperature dependence of electrical resistivity curves shows a semiconductor-like behavior (\({\rm d}\rho/{\rm d}T>0\)). At \(T<T_{\rm IM}\), it shows a metal-like behavior (\({\rm d}\rho/{\rm d}T>0\)). For the undoping sample \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\), its \( {T}_{\rm IM}\) is generally above 369 K [32]. Therefore, the substitution with magnetic ions decreases \(T_{\rm IM}\). Below \(T_{\rm IM}\), the resistivity decreases with the decrease in temperature. As we know, in the metallic conducting region, the metallic resistivity can be modeled by \(\rho _{\rm M}(T)=\rho _{0}+\rho _{2} {T}^{2}+\rho _{4.5} {T}^{4.5}+\rho _{5} {T}^{5}\) [33–36], where \(\rho _{0}\) is the residual resistivity, \( {T}^{2}\) is due to electron–electron scattering, \( {T}^{4.5}\) is due to electron–magnon scattering, \( {T}^{5}\) is due to electron–phonon scattering. In order to study the possible influence from magnetic ion of A-site, the electronic transport behavior in metallic region has been analyzed by the above formula. The metallic conductivity can be well fitted with this model from 285 K to the lowest temperature. The related fitting parameters are listed in Table 1. From it, one can find that with the decrease of average cationic radius and increase of cationic disorder, the electron–electron interaction decreases, while electron-phonon interaction increases. Generally, the ionic substitutions tend to decrease the conductivity in optimal \(\hbox {La}_{0.67}\hbox {Sr}_{0.33}\hbox {MnO}_{3}\) and \(\hbox {La}_{0.67}\hbox {Ca}_{0.33}\hbox {MnO}_{3}\). The main reason is due to the doping ions which narrow the energy band. In this situation, the decrease of carrier concentration will inevitably lead to a decline of electron–electron interaction in strength. On the other hand, the electron–phonon interaction will be enhanced due to the lattice distortion. Here, one can notice that, the scattering coefficients \(\rho _{4.5}\) are always negative in all doping samples, which are a non-physical behavior, indicating that the magnetic scattering need not be considered. Generally, the electron–magnon scattering is mainly due to the presence of spin fluctuation and spin disorder. In the current case, the Sm-, Dy-, and Er-doping inevitably influence the B-sited magnetic coupling and thus changes their spin structures. As the \(e_{\rm g}\) electrons hop through the \({\rm Mn}^{3+}{-}{\rm O}^{2-}{-}{\rm Mn}^{4+}\)transport channels, the induced spin inhomogeneities generate the electron–magnon interactions. However, considering the small doping concentration applied in the present samples, we think that the electron–magnon interaction should be limited. Combining the above analysis of magnetic behavior, we can clearly find that although the large magnetic moment ions of \(\hbox {Dy}^{3+}\) and \(\hbox {Er}^{3+}\) are doped into \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\), their magnetisms do not exhibit any obvious effects on electron transport. The properties can be understood from two aspects: first, the existence of A-site cation disorder makes the magnetic ions impossible to form an orderly arrangement; secondly, the doping magnetic ions are impossible to produce an effective magnetic coupling on each other due to the low doping concentration applied here. Thus, due to the lack of magnetic correlation on A-site, a single doping magnetic ion cannot change the exchange interaction of \(\hbox {Mn}^{3+}{-}\hbox {O}^{2-}{-}\hbox {Mn}^{4+}\) sublattice. As the large magnetic moment effects of doping ions are excluded, the lattice effect should play a key role in deciding the samples’ magnetic and electronic transport properties. Therefore, the decrease in Curie temperature and the increase of electron–phonon interaction mainly originate from the variation of average radius and cationic disorder on A-site.

Temperature T (K) dependence of resistivity \(\rho \) (\(\hbox {m}\Omega \,\hbox {cm}\)) for \(\hbox {La}_{0.7}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSMO), \(\hbox {La}_{0.6}\hbox {Sm}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSSMO), \(\hbox {La}_{0.6}\hbox {Dy}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSDMO), and \(\hbox {La}_{0.6}\hbox {Er}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (LSEMO). Red line represents the best fit to experimental \(\rho \)(T) curves by transport equation (Color figure online)
Conclusion
In summary, the IR spectra, Raman spectra, magnetization, and electrical resistivity of polycrystalline LSMO and Sm-(Dy, Er)-doping LSMO samples were reported. Two typical absorbed peaks of stretching mode \(\nu _{3}\) and bending mode \(\nu _{4}\) occur in the corresponding positions. However, in the Dy- and Er-doping samples, the peak of \(\nu _{3}\) reveals an obvious shift to low frequency region, indicating that the substitutions with Dy and Er strongly change Mn–O band lengths. All the JT modes almost appear at the same position exhibiting that the substitutions with different elements do not cause the variation of Mn–O–Mn band angles for all samples. The element substitution changes A-site average cationic radius and increases the disorder effect on A-site. Therefore, the Curie temperature \( {T}_{\rm C}\) was driven to low temperatures and the spin–cluster-glass state occurs in doping samples. The same isothermal magnetic behavior was observed at different temperatures (at 200 and 5 K) implying that B-site magnetic exchange interaction has not been influenced by the ionic substitution and even has nothing to do with the existence of A-site magnetism. In the study of \(\rho \)–T curves, the increase of electron–phonon interaction and decrease of electron–electron interaction also testify that the crystal lattice effect signally influences electrical transport. We suggest that, in low doping concentration, regardless of large or small magnetic dopant, the induced disorder effect and the variation of average cationic radius is a main factor to dominate the systematic magnetic–electronic properties.
References
von Helmolt R, Wecker J, Holzapfel B, Schultz L, Samwer K (1993) Giant negative magnetoresistance in perovskitelike \(\text{La}_{2/3}\text{Ba}_{1/3}\text{MnO}_{x}\) ferromagnetic films. Phys Rev Lett 71:2331–2333
Jin S, Tiefel TH, McCormack M, Fastnacht RA, Ramesh R, Chen LH (1994) Thousandfold change in resistivity in magnetoresistive La-Ca-Mn-O films. Science 264:413–415
Rao CNR, Cheetham AK (1999) Charge ordering in manganates. Science 276:911–912
Mori S, Chen CH, Cheong S-W (1998) Paired and unpaired charge stripes in the ferromagnetic phase of \(\text{La}_{0.5}\text{Ca}_{0.5}\text{MnO}_{3}\). Phys Rev Lett 81:3972–3975
Yunoki S, Hu J, Malvezzi AL, Moreo A, Furukawa N, Dagotto E (1998) Phase separation in electronic models for manganites. Phys Rev Lett 80:845–848
Mahendiran R, Tiwary SK, Raychaudhuri AK, Ramakrishnan TV, Mahesh R, Rangavittal N, Rao CNR (1996) Structure, electron-transport properties, and giant magnetoresistance of hole-doped \(\text{LaMnO}_{3}\) systems. Phys Rev B 53:3348–3358
Mahendiran R, Mahesh R, Raychaudhuri AK, Rao CNR (1996) Effect of Y substitution in La-Ca-Mn-O perovskites showing giant magnetoresistance. Phys. Rev. B 53:12160–12164
Sun Y, Xu XJ, Tong W, Zhang YH (2001) Tuning colossal magnetoresistance response by Cr substitution in \(\text{La}_{0.67} \text{Sr}_{0.33} \text{MnO}_{3}\). Appl Phys Lett 78:643–645
Dagotto E, Hotta T, Moreo A (2001) Colossal magnetoresistant materials: the key role of phase separation. Phys Rep 344:1–153
Rodriguez-Martinez LM, Attfield JP (1996) Cation disorder and size effects in magnetoresistive manganese oxide perovskites. Phys Rev B 54:R15622–15625
Louca D, Egami T, Brosha EL, Röder H, Bishop AR (1997) Local Jahn-Teller distortion in \(\text{La}_{1-x}\text{Sr}_{x}\text{MnO}_{3}\) observed by pulsed neutron diffraction. Phys Rev B 56:R8475–8478
Maignan A, Simon Ch, Caignaert V, Raveau B (1995) Giant magnetoresistance ratios superior to 1011 in manganese perovskites. Solid State Commun 96:623–625
Damay F, Martin C, Maignan A, Raveau B (1997) Cation disorder and size effects upon magnetic transitions in \(\text{Ln}_{0.5}\text{A}_{0.5}\text{MnO}_{3}\) manganites. J Appl Phys 82:6181–6184
Cao QQ, Wu J, Gu KM, Zhang SY, Du YW (1999) Large magnetostriction effect in bulk \((\text{La} _{1-x}\text{Sm} _{x})_{2/3}\text{Sr}_{1/3}\text{MnO}_{3}\). J Appl Phys 85:4494–4496
Liu YH, Wang CJ, Zhang RZ, Yue LQ, Luan KZ, Mei LM (2001) Substituting effects of Sm in polycrystalline La-Ca-Mn-O. J Phys Condens Matter 13:2009–2016
Mitra C, Raychaudhuri P, Dhur SK, Nigam AK, Pinto R, Pattalwar SM (1999) Evolution of transport and magnetic properties with dysprosium doping in \(\text{La}_{0.7-x}\text{Dy}_{x}\text{Sr}_{0.3}\text{MnO}_{3}\). J Magn Magn Mater 192:130–136
Bahadur D, Yewondwossen M, Kosiol Z, Foldeaki M, Dunlap RA (1996) Physical properties of the giant magnetoresistive perovskite system La-Er-Ca-Mn-O. J Phys Condens Matter 8:5235–5245
Sun Y, Salamon MB, Tong W, Zhang Y (2002) Magnetism, electronic transport, and colossal magnetoresistance of \((\text{La}_{0.7-x}\text{Gd}_{x})\text{Sr}_{0.3}\text{MnO}_{3} (0<\text{ x }< 0.6)\). Phys Rev B 66:094414–094419
Guo ZB, Huang H, Ding WP, Du YW (1997) Ferromagnetic state with spin canting in \(\text{La}_{0.52}\text{Gd}_{0.15}\text{Ca}_{0.33}\text{MnO}_{3}\). Phys Rev B 56:10789–10792
Yusuf SM, De Teresa JM, Ritter C, Serrate D, Ibarra MR, Yakhmi JV, Sahni VC (2006) Possible quantum critical point in \((\text{La}_{1-x}\text{Dy}_{x})_{0.7}\text{Ca}_{0.3}\text{MnO}_{3}\). Phys Rev B 74:144427–144432
Fan J, Ling L, Hong B, Zhang L, Pi L, Zhang Y (2010) Critical properties of the perovskite manganite \(\text{La}_{0.1}\text{Nd}_{0.6}\text{Sr}_{0.3}\text{MnO}_{3}\). Phys Rev B 81:144426–144431
Fan Jiyu, Ling Langsheng, Hong Bo, Pi Li, Zhang Yuheng (2009) Magnetocaloric effect in perovskite manganite \(\text{Nd}_{0.6}\text{La}_{0.1}\text{Sr}_{0.3}\text{MnO}_{3}\). J Magn Magn Mater 321:2838–2841
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr A 32:751–767
Hwang HY, Cheong SW, Radaelli PG, Marezio M, Batlogg B (1995) Lattice effects on the magnetoresistance in doped \(\text{LaMnO}_{3}\). Phys Rev Lett 75:914–917
Rodriguez-Martinez LM, Attfield JP (1998) Cation disorder and the metal-insulator transition temperature in manganese oxide perovskites. Phys Rev B 58:2426–2429
Nakamoto K (ed) (1986) Infrared and raman spectra of inorganic and coordination compounds, 4th edn. Wiley, New York, p. 148
Louca D, Egami T, Brosha EL, Röder H, Bishop AR (1997) Local Jahn-Teller distortion in \(\text{La}_{1-x}\text{Sr}_{x}\text{MnO}_{3}\) observed by pulsed neutron diffraction. Phys Rev B 57:R8475–8482
Iliev MN, Abrashev MV, Popov VN, Hadjiev VG (2003) Role of Jahn-Teller disorder in Raman scattering of mixed-valence manganites. Phys Rev B 67:212301–212304
Pollert E, Jirák Z, Hejtmánek J, Strejc A, Kužel R, Hardy V (2002) Detailed study of the structural and magnetic transitions in \(\text{Pr}_{1-x}\text{Sr}_{x}\text{MnO}_{3}\) single crystals (\(0.48\le \text{x }\le 0.57\)). J Magn Magn Mater 246:290–296
Ahn KH, Wu XW, Liu K, Chien CL (1996) Magnetic properties and colossal magnetoresistance of \(\text{La(Ca)MnO}_{3}\) materials doped with Fe. Phys Rev B 54:15299–15302
Rubinstein Mark, Gillespie DJ, Snyder John E, Tritt Terry M (1997) Effects of Gd Co, and Ni doping in \(\text{La}_{2/3}\text{Ca}_{1/3}\text{MnO}_{3}\) : resistivity, thermopower, and paramagnetic resonance. Phys Rev B 56:5412–5423
Urushibara A, Moritomo Y, Arima T, Asamitsu A, Kido G, Tokura Y (1995) Insulator-metal transition and giant magnetoresistance in \(\text{La}_{1-x}\text{Sr}_{x}\text{MnO}_{3}\). Phys Rev B 51:14103–14109
Li R-W, Zhou X, Shen BG, Hillebrands B (2005) Process dependence of transport properties in phase-separated \(\text{Pr}_{ 0.7}\text{Pb}_{0.3}\text{MnO}_{3}\) single crystals. Phys Rev B 71:092407–092410
Schiffer P, Ramirez AP, Bao W, Cheong SW (1995) Low temperature magnetoresistance and the magnetic phase diagram of \(\text{La}_{1-x}\text{Ca}_{x}\text{MnO}_{3}\). Phys Rev Lett 75:3336–3339
Snyder GJ, Hiskers R, Dicarolis S, Beasley MR, Geballe TH (1996) Intrinsic electrical transport and magnetic properties of \(\text{La}_{0.67}\text{Ca}_{0.33}\text{MnO}_{3}\) and \(\text{La}_{0.67}\text{Sr}_{0.33}\text{MnO}_{3}\) MOCVD thin films and bulk material. Phys Rev B 53:14434–14444
Li G, Zhou HD, Feng SJ, Fan XJ, Li XG, Wang ZD (2002) Competition between ferromagnetic metallic and paramagnetic insulating phases in manganites. J Appl Phys 92:1406–1411
Acknowledgements
This work was supported by the funding of Jiangsu Innovation Program for Graduate Education (Grant No. KYZZ-0090) (Fundamental Research Funds for the Central Universities) and the National Nature Science Foundation of China (Grant Nos. 11475086, 51202235, 11204131 and U1332140)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fan, J., Xu, L., Zhang, X. et al. Effect of A-site average radius and cation disorder on magnetism and electronic properties in manganite \(\hbox {La}_{0.6}\hbox {A}_{0.1}\hbox {Sr}_{0.3}\hbox {MnO}_{3}\) (A = Sm, Dy, Er). J Mater Sci 50, 2130–2137 (2015). https://doi.org/10.1007/s10853-014-8775-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8775-1