
Abstract
In this work, the effects of changing molecular weight of polyol (2000, 3000, and 4000) and block ratio as well as deformation amplitude on thermal, structural, and shape memory properties of polyester urethanes based on diphenylmethane diisocyanate, polycaprolactone diol, and 1,4-butanediol were investigated. Fourier transform infrared spectroscopy was used to check the accomplishment of the polyurethanes synthesis. Thermal, structural, and shape memory properties of synthesized SMPUs were measured using differential scanning calorimetry, wide angle X-ray diffraction, and tensile cyclic tests, respectively. It was found that as the crystallinity of soft segments increased, the ability of the samples in fixation of temporary shape was higher. On the other hand, the shape recovery was dominated by the hard segment content and there was a minimum critical HSC for the samples to show appropriate shape memory effects.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Shape memory polymers (SMPs) are a category of smart materials, with the ability to keep the deformed shape at the service temperature and revert back to the original shape by the application of an external stimuli, such as light, humidity, radiation, heat or electrical, and magnetic field [1–4]. However, a majority of these materials are stimulated by the application of heat. The change in shape which is caused by heating is called thermally induced shape memory effect (SME) [5]. This type of SME has been found in various materials among which the shape memory alloys and polymers are the most popular [6, 7].
Shape memory polyurethanes (SMPUs) have gained widespread attention in biomedical (such as orthodontics, stents, and sutures) [8–10], industrial, and academic applications due to light weight, low cost, easy processing, and high shape recovery strain [11, 12]. In case of medical usage, these polymers are expected to be biodegradable and have shape recovery temperature near the human body temperature [13]. Basically, these SMPUs are consisted of two phases, the frozen phase and the reversible phase [14, 15]. In some cases, soft domain crystals form the reversible phase [16] with their crystalline melting temperatures being the shape recovery temperature (T r), while the hard domains constitute the frozen phase [17]. Generally, SMPU is a stimuli-sensitive block copolymer, having the ability to change its shape at a temperature above a switching temperature (T s) that could be either the glass transition or the melting temperature of the soft segment domain [18]. This process consists of an elastic deformation by applying an external stress and subsequent temporary fixation. Generally, the crystallinity of soft segments and the formation of stable hard segment domains, acting as physical crosslink points in the temperature range above the melting temperature of soft segment crystals (T m.s), are the two essential requirements for polyurethanes each having a melting switching temperature [19]. Basically, there are two major synthetic parameters that control the shape memory properties, the hard segment content (HSC), and the soft segment length (SSL). Thermomechanical cyclic conditions can also enormously affect the shape memory properties [20]. Several factors such as phase separation, phase composition, micro-domain size, and phase distribution dictate the structure of physically cross-linked networks and shape memory properties of the SMPUs [21, 22]. The relationship between morphology and shape memory properties is essential for optimizing the molecular design and applications of SMPUs.
Lee et al. [23] studied the structure and thermomechanical properties of polyether-based SMPUs. They synthesized polyurethanes composed of 4,4′-methylenediphenyl diisocyanate (MDI), polytetramethylene ether glycol (PTMEG), and 1,4-butanediol (1,4-BDO) and investigated the effect of HSC on their properties. The authors reported that 30–45 wt% of HSC resulted in 80–95 % of shape recovery and the HSC was an essential parameter in determining the physical properties of PU block copolymers. Ji et al. [24] investigated the morphology and SME of segmented polyurethanes with crystalline reversible phase. They synthesized a set of segmented polyurethanes composed of MDI, 1,4-BDO, and polycaprolactone diol (PCL-diol) with M w = 4000 having different HSCs (10–50 wt%) and showed that the samples had either an interconnected, isolated, or no hard-segmented domains. These authors have also mentioned that the shape fixity of the segmented polyurethane samples reduced with the increase of HSC and the samples with isolated hard segment domains showed better shape recoveries than that of interconnected ones.
In addition, the thermomechanical conditions in terms of deformation temperature, time, and speed could greatly influence the shape memory behaviors [25]. The segmented polyurethanes with different structures should also show different property dependency on the thermomechanical conditions. Tobushi et al. [26] studied the effect of strain-holding temperature and strain-holding time on shape recovery and the irrecoverable strain control of a special polyurethane film produced by Mitsubishi Heavy Industries Ltd. They found that the higher shape-holding temperature and the longer shape-holding time resulted in the higher irrecoverable strain rate.
In this work, a series of polycaprolactone-based SMPUs with different SSLs and various HSCs were synthesized using PCL-diol (with different molecular weights), MDI and 1,4-BDO by bulk polymerization method. In order to check the accomplishment of the polyurethanes synthesis all the samples were examined by a Fourier transform infrared (FT-IR) spectroscope. Thermal and structural properties were evaluated using differential scanning calorimetry (DSC) and wide angle X-ray diffraction (WAXD) techniques, respectively. The tensile and shape memory properties of synthesized SMPUs were also examined by thermomechanical cyclic tests. In addition, the effect of strain amplitude on shape memory properties of specimens was investigated.
Experimental
Materials
PCL-diols with average molecular weight of 2000, 3000, and 4000 (namely CAPA2000, CAPA3000, and CAPA4000) were supplied by Solvay Interox and dried at 80 °C under vacuum for 24 h before synthesis. Extra pure grade of MDI was supplied by Aldrich and used to synthesize the samples without further treatment. Dimethylformamide (DMF) and 1,4-BDO were both supplied by Merck and dried by using appropriate molecular sieves for several days before use.
Synthesis of SMPUs
Three series of thermoplastic polyurethanes (TPUs) with different SSLs and HSCs (resulting from different block ratios) were synthesized by the pre-polymerization method. A 500-ml, round-bottom, four-necked separable flask equipped with a mechanical stirrer, a nitrogen inlet, a thermometer, and a condenser with a drying tube was used as a reactor. TPUs were synthesized by bulk polymerization in DMF under dry N2 in two steps. An appropriate amount of MDI and PCL-diol in 100 cm3 of DMF, which was freshly distilled before use, were stirred under nitrogen at 80 °C for 2 h to make the prepolymer. 1,4-BDO was then added drop-wise to the reaction mixture according to the selected MDI/PCL ratios. The reaction mixture became very sticky upon polymerization, and it was carried out until the unreacted isocyanate groups could not be observed by FT-IR. After polymerization, the prepared samples were all vacuumed in order to remove the air bubbles and then dried by storing in an oven at 100 °C for 24 h. Formulations of the samples are presented in Table 1. The structure of SMPUs with various SSL and HSC are also illustrated in Scheme 1. As it has been shown, the increase of block ratio results in the development of more dense hard domains and the increase of PCL-diol molecular weight causes the production of longer soft domains, resulting in the formation of soft crystalline regions.

The structure of polyurethane and its evolution with the change in SSL and HSC
Characterization
FT-IR spectroscopy was used on a Bruker Equinox 55 instrument in order to identify the urethane linkage and phase separation. Thermal behavior of 5 ± 0.1 mg of each sample was determined by using a DSC instrument (Perkin-Elmer) under the flow of nitrogen gas. All the samples were first heated from −80 to 220 °C at a heating rate of 10 °C/ min, held at 220 °C for 1 min, subsequently, cooled to −80 °C at a cooling rate of 10 °C/min , held at −80 °C for another 1 min and then reheated to 220 °C at the same heating rate. WAXD was performed on a Siemens, D5000 X-ray diffractometer using Cu Kα radiation (λ = 1.54 Å) at a scan rate of 1°/min. Cyclic tensile tests were performed on an Instron 6022 apparatus equipped with a temperature-controlled chamber. Each sample with the dimensions of 20 × 2 × 0.5 mm3 was first heated up to the temperature of 70 °C (T high) in 600 s. It was then stretched to the maximum elongation (ɛ m) of 50 and 100 % with the strain rate of 0.5 min−1 at T high. Nitrogen was then purged into the chamber to decrease the temperature to −20 °C (T low), in order to freeze the sample while it was held at the constant strain, ɛ m. Subsequently, the strain was released from ɛ m to zero and reheating started for another 600 s. The cycle was repeated three times for each sample to evaluate the SME. The shape fixity (R f(N)) and shape recovery (R r(N)) after the Nth cycle were both calculated using the following equations [24, 27, 28]:
where ɛ m is the maximum strain applied to the sample, ɛ u(N) is the strain of the sample right after unloading at the Nth cycle, and ɛ p(N) is the recorded strain at the Nth cycle in thermo-mechanical analysis. Shape fixity represents the ability of a sample to keep its temporary shape and shape recovery shows how SMPU sample can return to its original shape [24]. Mechanical properties were investigated through the thermomechanical cyclic tests. No independent mechanical properties measurements were carried out in this case and the maximum tensile stress in each cycle was taken as the strength of each specimen.
Results and discussion
Spectroscopic characterization
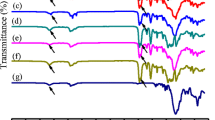
Figure 1 shows the FT-IR spectra of three samples with different HSC and SSL. As it can be seen, the disappearance of –NCO absorption peaks at 2270 cm−1 and the presence of two peaks at 1730 and 3343 cm−1 due to C=O and –NH groups, respectively, are in agreement with the formation of urethane bonds. The FT-IR spectra of the polyurethanes exhibited typical peaks at 3343, 2930, and 1070 cm−1 corresponding to –NH, CH2, and –C–O–C–, respectively [29, 30]. Two characteristic peaks observed at about 1710 and 1730 cm−1 are related to the stretching vibration of carbonyl groups (C=O) in hard segments. The former peak is due to the presence of hydrogen bonded carbonyl groups of urethane formed by phase separation and intermolecular interaction with –NH in hard segments, whereas the latter peak is due to the presence of symmetrical stretching of non-hydrogen bonded carbonyl group due to dissolving in the matrix of soft segments [23, 31, 32]. The intermolecular hydrogen bonding between C=O and –NH group in the hard segment is responsible for phase separation of soft and hard domains. It should be mentioned that the degree of phase separation is partially under the control of HSC. As HSC increases, the degree of phase separation also increases proportionally, which implies the enhancement of interaction among hard segments through hydrogen bonding. In addition, new absorption bands (amide II) at 1530 cm−1 for MDI-based polyurethanes are also observed. The intensities of these new bands also increase with the increase of HSC [33].

Typical FT-IR spectra of SMPUs: a 4000-33, b 3000-39, and c 2000-50
Thermal analysis
Figures 2, 3, and 4 show the second heating scans of 2000, 3000, and 4000 samples. The DSC data obtained from the figures have also been presented in Table 2. As it can be observed, contrasting to 3000 and 4000 samples, no crystalline structure of soft segments was observed in 2000 series. This could be attributed to the lower molecular weight of PCL-diol used in 2000 series in comparison with the two other series which prevent the former to form well-developed phase separated regions and thus, show more phase mixing. It can also be concluded that the low molecular weight of CAPA2000 prevent the polymeric chains to form crystalline structures. For 2000-44 and 2000-50, two endothermic peaks are observed at 189.2 and 196.8 °C, respectively, that are related to the melting of hard domains (Fig. 2). It is clear from the figure that the melting point of hard domains (T m.h) is increased with the increase of HSC. In the temperature range −60 to −30 °C, a change in the baseline of all samples is observed due to the glass transition temperature of soft segments (T g.s). Interconnections between hard and soft segments are improved with increasing of HSC due to the formation of hydrogen bonding between the hard domains. Therefore, the mobility of soft domains is restricted in the presence of hard domains, which results in the increasing of glass transition temperature of soft segments.

DSC heating scans of 2000 samples: a 2000-32, b 2000-39, c 2000-44, and d 2000-50

DSC heating scans of 3000 samples: a 3000-24, b 3000-30, c 3000-35, and d 3000-39

DSC heating scans of 4000 samples: a 4000-19, b 4000-24, c 4000-29, and d 4000-33
It can also be seen from Figs. 3, 4, and Table 2 that the enthalpy of melting (ΔH m.s) and melting point (T m.s) of soft segments decrease slightly with the increase of HSC for each 3000 and 4000 series. The degree of crystallinity (X c) of soft segments can be calculated from the ratio of the enthalpy of melting for the soft segment phase to that of the pure PCL. It can be observed that at the same block ratios for the 3000 and 4000 series, ΔH m.s is also increased with the increase in PCL-diols chain lengths due to the higher degree of crystallinity of soft segments. Moreover, longer PCL-diols chain lengths will increase the distance between the hard and soft segments, leading to the formation of much stronger soft domains. Comparing Figs. 2, 3, and 4, it could be observed that the heat of fusion of the melting peaks which are seen at about 197 °C are decreased with the increase in SSL. These peaks are attributed to the melting of hard domains. In this case, it would be difficult for hard segments to form large domains; therefore, the number of hydrogen bonds formed would be less in this case.
The cooling scans of 2000 and 3000 samples are shown in Figs. 5 and 6. The existence of crystallization peaks are related to the hard domains and segments. Figure 5 shows that with the increase of HSC, the crystallization peak temperature (T c) also shifts to the higher temperatures. The crystallization enthalpy and temperature of all samples are also presented in Table 2. Because of low HSC, no ordered and strong domains are formed and the broad peak between the two separated peaks can be attributed to the arrangement of dispersed and disordered hard segments. This behavior can be seen for 2000-32 and 3000-24 samples. The crystallization peaks for 4000 samples are shown in Fig. 7. For all samples, strong crystalline peaks are observed at −6 to 13 °C. Comparing Figs. 5, 6, and 7, it can be observed that as the molecular weight of polyol reduces from 4000 to 2000, the formation of hard domains will be more intensified. It can be observed that the enthalpy of hard segments crystallization (ΔH c.h) and also their crystallization temperatures (T c.h) increase generally with the increase of HSC for each series of samples synthesized. For 4000 samples, the enthalpy of soft segments crystallization (ΔH c.s) and their crystallization temperatures (T c.s) decreased with the increase of HSC. This could be attributed to the effect of hard segments on PCL-diol chains. These results implied that the crystallinity of soft segments were influenced by HSC and SSL, in which it was decreased and increased with the increase of the former and latter, respectively.

DSC cooling scans of 2000 samples: a 2000-32, b 2000-39, c 2000-44, and d 2000-50

DSC cooling scans of 3000 samples: a 3000-24, b 3000-30, c 3000-35, and d 3000-39

DSC cooling scans of 4000 samples: a 4000-19, b 4000-24, c 4000-29, and d 4000-33
Crystalline structure
Typical WAXD patterns of the samples prepared from different series are shown in Fig. 8. As it can be seen, no peak is observed for the 2000 samples, indicating that no crystalline structure is present in the soft segments of the 2000 samples. This is also consistent with the DSC results. It is also observed that all the samples with SSL of 3000 and 4000 have two prominent peaks at about 2θ of 21° and 23°, indicating that the crystalline structure stems from the PCL soft segment [34]. It is clear that the intensity of the two peaks decreases with increasing of HSC. This is in agreement with the results reported by Kim and Lee [35] that the two peaks are related to the crystallization of soft segments. In other words, WAXD can only show the crystalline regions of soft segments. Table 3 represents a comparison between the results of DSC and WAXD for 4000 series. As it can be seen, there is not much difference between the data obtained from the two different methods.

WAXD patterns of 2000, 3000 and 4000 series with different hard segment content: a 4000-19, b 4000-24, c 3000-24, d 3000-30, e 2000-32, and f 2000-39
In contrast to the soft segment crystals, the peak related to the hard domains can be characterized using small angle X-ray scattering (SAXS) technique. Pongkitwitoon et al. [36] studied MDI/1,4-BDO hard blocks of polyurethanes with various soft segments. It was revealed that the peak attributed to the hard segments was emerged at the scattering angle of about 0.7°. They also reported that the peak was shifted to the higher scattering angles with the increase of temperature. Linliu et al. [37] synthesized polyurethanes based on MDI, 1,4-BDO, and polyester diols with HSC of 48 %. They showed that the peak was appeared at the scattering angle of 0.4°. According to other published works [38, 39], it can be concluded that the peak attributed to MDI/l,4-BDO hard domains is generally emerged at the scattering angles of less than 1°.
Shape memory properties
In addition to synthetic parameters such as HSC and SSL, thermomechanical cyclic conditions like the strain amplitude could also affect the shape memory properties [19, 25, 26, 33]. Figure 9a–d shows the thermomechanical cycles for the samples prepared using CAPA2000 with different block ratios. There is a notable difference between the first and second cycle [28] in each sample which could be attributed to the irreversible disentanglement of the polymeric chains in the first elongation [6]. Table 4 represents the shape memory properties of the samples in both 50 and 100 % elongation. Comparing the samples with equal block ratios, it can be seen that the higher degree of crystallinity of soft segments results in an improved shape fixity. Table 4 indicates that R f increases with the increase of SSL and the maximum and minimum amounts of R f are observed for 4000-24 and 2000-50 samples, respectively. In contrast, shape recovery is controlled by HSC [23] and as it is clear from the table, the highest and lowest values have been obtained for 2000-50 and 4000-24, respectively. No R f and R r values have been reported for the 4000-19 sample in Table 4. In fact, this sample was broken at less than 50 % of elongation at high temperature. Therefore, no data could be presented here. As it was mentioned before, hard domains are responsible for the recovery of samples to the original shape. The behavior of 4000-19 sample during deformation revealed that a minimum HSC is required to form the strong hard segments and domains. Thus, it can be concluded that a higher amount of HSC rather than 19 wt% should have been selected for 4000 series in order to make a material with a suitable shape memory behavior.

Thermomechanical cyclic tensile tests for the 2000 series: a 2000-32, b 2000-39, c 2000-44, and d 2000-50
The effect of strain amplitude on shape fixity of all specimens is shown in Fig. 10a and b. As it can be observed, R f increases dramatically with the increase in strain amplitude which may be due to the orientation of soft segments in higher elongations. As the strain amplitude increases, the mobility of soft segment chains are more restricted, and consequently they tend to preserve their previous chain conformations. Figure 11a and b shows the effect of strain amplitude on R r of the samples. The R r value decreases with the increase of strain amplitude. This can be attributed to the disentanglement of polymeric chains of the samples which result in irreversible deformations. Comparing Figs. 10 and 11, it can be concluded that R f and R r show contradictory reactions upon deformation. Therefore, it is crucial to apply a suitable elongation to obtain optimum shape memory properties. The figures also reveal that the variation of R f and R r could be approximated linear in the range studied.

Effect of strain amplitude on shape fixity of the selected specimens in a 50 % elongation and b 100 % elongation

Effect of strain amplitude on shape recovery of the selected specimens in a 50 % elongation and b 100 % elongation
Comparing sample 4000-29 with a similar reported one (sample IIc) in the literature [11], it is observed that at the same soft segment content and strain amplitude, the shape recovery and shape fixity of our sample has been improved and deteriorated, respectively. Despite the fact that sample IIc was chemically crosslinked, the increase in the shape recovery of 4000-29 could be attributed to the higher shape recovery temperature, while the reduced shape fixity could be related to the absence of chemical crosslinks. In addition, PCLU-M sample synthesized by Ping et al. [21] showed a relatively higher shape recovery (95.8 %) in comparison with 4000-33 sample (93.5 %) in 100 % elongation. This can mostly be attributed to the different block ratios selected in the two different studies. It should also be anticipated that in the case of using higher block ratio, our sample could have shown a higher shape recovery than that of the value reported.
Our studies on the shape recovery and shape fixity of the 2000 samples can also be compared with those having poly(tetramethylene glycol) as the soft segments [23]. It is found that for approximately the same HSCs (23, 39, and 44), the shape recovery values of our samples are higher, however, the shape fixity values are lower. The higher shape recovery can be attributed to the presence of more hydrogen bondings in the system. It is worth to mention that more hydrogen bondings are formed in ester-based PUs in comparison with ether-based ones. On the other hand, the deterioration of shape fixities of our samples can be related to their shape fixity temperatures. Although, the degree of crystallinities of ester-based PUs are higher than those of ether-based PUs, the lower fixity temperature of sample containing poly(tetramethylene glycol) is responsible for its better shape fixity. Therefore, the shape fixity and shape recovery are dependent on SSL and HSC, respectively, and by selecting proper values of these two, along with suitable strain amplitude, optimum shape memory properties could be obtained.
Maximum tensile stress of the samples at typically 50 % elongation after three cycles is illustrated in Table 5. The table shows that the maximum tensile stress is dramatically increased with increasing of HSC for each series of samples. This implies the fact that HSC also plays an important role in mechanical properties. With increasing of HSC, independent domains of hard segments are formed and more phase separation between hard and soft segments is taken place. Therefore, more hydrogen bonds are created within the hard domains in this case, which results in higher tensile strengths for the SMPU samples.
Conclusions
Three series of segmented polyurethanes were synthesized using PCL-diols with different molecular weights, MDI and 1,4-BDO by bulk polymerization method. The FT-IR spectra indicated that the urethane linkage was appeared and also isocyanate peaks related to the MDI were disappeared for all the samples. These both are in agreement with the accomplishment of the synthesis of the polyurethanes prepared. DSC and WAXD data showed that a minimum SSL was required for the formation of crystalline structures in the polyols. Thus, no crystalline structure of the soft segments was observed in the 2000 series, whereas 3000 and 4000 series contained crystalline soft domains. The results revealed that beyond a critical amount of HSC, crystalline hard domains of MDI and 1,4-BDO were formed. This was noticed by DSC at higher temperatures. The effects of HSC, SSL, and strain amplitude on shape memory properties of the samples were also investigated. For all specimens, shape recovery and shape fixity values were increased and decreased, respectively, with the increase of HSC. It was also obtained that the shape fixity is dominated by the crystallization of soft segments, whereas the shape recovery is under the control of HSC. In other words, the samples with higher SSL showed better shape fixity and the specimens containing higher HSC showed better shape recovery. It could also be concluded that in order to have optimum shape memory properties, selecting of an optimum amount of SSL and HSC is necessary. The samples showed higher shape fixity and lower shape recovery with the increase of strain amplitude. The former was due to the possible strain induced crystallization in the soft segment regions, while the latter could be related to the irreversible disentanglements and possible damages occurred in the polymeric chains.
References
Sahoo NG, Jung YC, Yoo HJ, Cho JW (2007) Influence of carbon nanotubes and polypyrrole on the thermal, mechanical and electroactive shape-memory properties of polyurethane nanocomposites. Compos Sci Technol 67:1920
Liu Y, Gall K, Dunn ML, McCluskey P (2003) Thermomechanical recovery couplings of shape memory polymers in flexure. Smart Mater Struct 12:947
Li J, Viveros JA, Wrue MH, Anthamatten M (2007) Shape memory effects in polymer networks containing reversibly associating side-groups. Adv Mater 19:2851
Gunes IS, Cao F, Jana SC (2008) Evaluation of nanoparticulate fillers for development of shape memory polyurethane nanocomposites. Polymer 49:2223
Gunes IS, Cao F, Jana SC (2008) Effect of thermal expansion on shape memory behavior of polyurethane and its nanocomposites. J Polym Sci B 46:1437
Meng Q, Hu J, Zhu Y, Lu J, Liu Y (2007) Polycaprolactone-based shape memory segmented polyurethane fiber. J Appl Polym Sci 106:2515
Liu Y, Chung A, Hu J, LV J (2007) Shape memory behavior of SMPU knitted fabric. J Zhejiang Univ Sci A 8:830–834
Jung YC, Cho JW (2010) Application of shape memory polyurethane in orthodontic. J Mater Sci Mater Med 21:2881. doi:10.1007/s10856-008-3538-7
Wache HM, Tartakowska DJ, Hentrich A, Wagner MH (2003) Development of a polymer stent with shape memory effect as a drug delivery system. J Mater Sci Mater Med 14:109. doi:10.1023/A:1022007510352
Weiss RA, Izzo E, Mandelbaum S (2008) New design of shape memory polymers: mixtures of an elastomeric ionomer and low molar mass fatty acids and their salts. Macromolecules 41:2978
Hu J, Yang Z, Yeung L, Ji F, Liu Y (2005) Crosslinked polyurethanes with shape memory properties. Polym Int 54:854
Yang B, Huang WM, Li C, Lee CM, Li L (2004) On the effects of moisture in a polyurethane shape memory polymer. Smart Mater Struct 13:191
Ping P, Wang W, Chen X, Jing X (2005) Poly(ε-caprolactone) polyurethane and its shape-memory property. Biomacromolecules 6:587
Barikani M, Zia KM, Bhatti IA, Zuber M, Bhatti HN (2008) Molecular engineering and properties of chitin based shape memory polyurethanes. Carbohydr Polym 74:621
Zhu Y, Hu J, Yeung LY, Liu Y, Ji F, Yeung KW (2006) Development of shape memory polyurethane fiber with complete shape recoverability. Smart Mater Struct 15:1385
Gunes IS, Jimenez GA, Jana SC (2009) Carbonaceous fillers for shape memory actuation of polyurethane composites by resistive heating. Carbon 47:981
Zia KM, Zuber M, Barikani M, Bhatti IA, Khan MB (2009) Surface characteristics of chitin-based shape memory polyurethane elastomers. Colloid Surf B 72:248
Chen S, Hu J, Yuen CW, Chan L (2009) Supramolecular polyurethane networks containing pyridine moieties for shape memory materials. Mater Lett 63:1462
Chen S, Hu J, Liu Y, Liem H, Zhu Y, Liu YJ (2007) Effect of SSL and HSC on morphology and properties of PHA based SMPU synthesized by bulk polymerization method. J Polym Sci B 45:444
Hu JL, Ji FL, Wong YW (2005) Dependency of the shape memory properties of a polyurethane upon thermomechanical cyclic conditions. Polym Int 54:600
Ping P, Wang W, Chen X, Jing X (2007) The influence of hard-segments on two-phase structure and shape memory properties of PCL-based segmented polyurethanes. J Polym Sci B 45:557
Meng Q, Hu J, Mondal SJ (2008) Thermal sensitive shape recovery and mass transfer properties of polyurethane/modified MWNT composite membranes synthesized via in situ solution pre-polymerization. Membr Sci 319:102
Lee BS, Chun BC, Chung YC, Sul KII, Cho JW (2001) Structure and thermomechanical properties of polyurethane block copolymers with shape memory effect. Macromolecules 34:6431
Ji FL, Hu JL, Li TC, Wong YW (2007) Morphology and shape memory effect of segmented polyurethanes. Part I: With crystalline reversible phase. Polymer 48:5133
Tobushi H, Hayashi S, Hoshio K, Miwa N (2006) Influence of strain-holding conditions on shape recovery and secondary-shape forming in polyurethane-shape memory polymer. Smart Mater Struct 15:1033
Tobushi H, Hayashi S, Hoshio K, Ejiri Y (2008) Shape recovery and irrecoverable strain control in polyurethane shape-memory polymer. Sci Technol Adv Mater 9:015009
Kang SM, Lee SJ, Kim BK (2012) Shape memory polyurethane foams. Express Polym Lett 6:63
Leonardi AB, Fasce LA, Zucchi IA, Hoppe CE, Soule ER, Perez CJ, Williams RJJ (2011) Shape memory epoxies based on networks with chemical and physical crosslinks. Eur Polym J 47:362
Raja M, Ryu SH, Shanmugharaj AM (2013) Thermal, mechanical and electroactive shape memory properties of polyurethane (PU)/poly (lactic acid) (PLA)/CNT nanocomposites. Eur Polym J 49:3492
Oprea S, Oprea V (2010) Influence of crosslinkers on properties of new polyurethane elastomers. Mater Plast 47:54
Yeganeh H, Razavi-Nouri M, Ghaffari M (2008) Synthesis and properties of polybenzoxazine modified polyurethanes as a new type of electrical insulators with improved thermal stability. Polym Eng Sci 48:1329
Yeganeh H, Razavi-Nouri M, Ghaffari M (2008) Investigation of thermal, mechanical, and electrical properties of novel polyurethanes/high molecular weight polybenzoxazine blends. Polym Adv Technol 19:1024
Wang W, Ping P, Chen X, Jing X (2007) Shape memory effect of poly(l-lactide)-based polyurethanes with different hard segments. Polym Int 56:840
Sivakumar C, Nasar AS (2009) Poly(ε-caprolactone)-based hyper branched polyurethanes prepared via A2 + B3 approach and its shape-memory behavior. Eur Polym J 45:2329
Kim BK, Lee SY (1996) Polyurethanes having shape memory effects. Polymer 37:5781
Pongkitwitoon S, Hernandez R, Weksler J, Padsalgikar A, Choi T, Runt J (2009) Temperature dependent microphase mixing of model polyurethanes with different intersegment compatibilities. Polymer 50:6305
Linliu K, Chen SA, Yu TL, Lin TL, Lee CH, Kai JJ, Chang SL, Lin JS (1995) A small-angle X-ray scattering study of microphase separation transition of polyurethanes: effect of hard segments. J Polym Res 2:63
Koberstein JT, Stein RS (1983) Small-angle X-ray scattering studies of microdomain structure in segmented polyurethane elastomers. J Polym Sci B 21:1439
Chu B, Li Y (1993) Synchrotron SAXS studies of segmented polyurethanes. Progr Colloid Polym Sci 91:51
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Momtaz, M., Razavi-Nouri, M. & Barikani, M. Effect of block ratio and strain amplitude on thermal, structural, and shape memory properties of segmented polycaprolactone-based polyurethanes. J Mater Sci 49, 7575–7584 (2014). https://doi.org/10.1007/s10853-014-8466-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8466-y