
Abstract
For the first time, pure protein fibers with good mechanical properties were obtained from waste wool after effective de-crosslinking and disentanglement of the highly crosslinked keratin. Every year, more than 1.6 billion pounds of wool keratin-based materials were discarded. Effort has been devoted to convert the keratin resources into high value-added products, especially fibers. In 1940s, pure keratin fibers had been developed from feathers. However, after trying the method, we found the results were not repeatable and did not find any other successful repetition. So far, no effective dissolution and spinning methods have been developed to obtain pure keratin fibers with potential for real applications. In this research, keratin with preserved backbones after de-crosslinking was obtained. Subsequently, surfactant endowed the keratin with satisfactory stretchability for fiber spinning. Fibers of 20 μm with good tensile strength were successfully developed. The technology could be applied onto other highly crosslinked proteins, including feather keratin from poultry industry, sorghum protein and soy protein in bioenergy co-products for production of various industrial products.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fiber industries are encountering problems of shortage and non-sustainability of resources [1]. The global fiber production of 85.8 million tons in 2012 mainly consisted of about 59 % of oil-derived synthetic fibers and 35 % of cotton [2]. The increasing world demand for fibers could not be satisfied, since the petroleum resource is depleting, while the production of cotton is not increasing [3]. Moreover, dependence on petroleum remains another obstacle for sustainable development of fiber industries. Therefore, it is urgent to seek low-cost renewable raw materials for sustainable fiber production.
Waste wool could meet requirements for fiber spinning in terms of both availability and technical feasibility. Waste wool could be mainly obtained from textile processing, discarded wool textiles, meat goats, and sheep. First, wool textile processing, especially scouring, carding, combing, yarn spinning, generated large quantities of waste wool that were too short for direct textile processing [4]. Secondly, about half of the 490 million pounds of used wool textiles were discarded as municipal solid wastes, and ended up in incineration or landfill [4, 5]. Thirdly, the global meat goats and sheep annually generated up to 1.1 billion pounds of short and coarse fibers, which usually could not be fabricated into high value-added products in textiles [6]. Wool contains about more than 80 wt% of keratin with molecular weight higher than 10 kDa, which was the premise for fiber spinning [7].
Much research has been done on converting waste wool and other keratin-based wastes into valuable materials [6]. Waste keratin has been pulverized into powders for further processing [8, 9], developed into films and lightweight composites [10, 11], chemically modified into thermoplastics and adsorbents [12, 13], or converted into textile sizes [14]. However, fiber spinning was a less studied approach. Spinning of fibers had much higher standards in terms of molecular weights, linearity of molecules and drawability of the solution than production of films and other non-fibrous structures.
To the best of our knowledge, no effective method has been developed to produce regenerated keratin fibers. In 1940s, several reports had been published on developing pure keratin fibers from chicken feathers via wet spinning [15–17]. However, mechanical properties of the fibers were not reported. Nevertheless, we could neither repeat the results from the experiments, nor find any other reported successful repetition of the methods. In 2000s, U.S. Department of Agriculture had investigated funds on converting chicken feathers into industrial products, including fibers [18, 19]. However, no report regarding successful production of keratin fibers were found. The latest report regarding spinning of pure regenerated keratin fibers was via ionic liquid dissolution in 2008 [20]. Fan dissolved feather keratin in ionic liquid for wet spinning [21]. However, the obtained fibers showed tensile strength as low as 0.2 g/denier. In addition, keratin has been spun into fibers via blending with polyethylene oxide [22], cellulose acetate [23], cellulose [24], silk [25, 26], poly-3-hydroxybutyrate-co-valerate [27], etc., as well as extruded with plasticizers [28, 29]. So far, imparting sufficient spinnability to pure keratin remained challenging.
To solve this problem, suitable dissolution methods should be used to disentangle and align the linear keratin molecules. Current dissolution methods were summarized here. Alkaline treatment randomly destroyed backbones and disulfide bonds in keratin, and resulted in short molecules that could not be spun solely [30]. Extraction of feather keratin with thiol could maintain the molecular backbones while dissociating the disulfide crosslinks. However, fibers could not be fabricated if the extracted linear keratin remained highly entangled in solution [31]. Moreover, most widely used thiols, such as mercaptoethanol and dithiothreitol, were either harmful to the environment or too expensive for large-scale use. Sodium sulfites could also reduce keratin, but might have lower yield than thiols, such as cysteine [32]. In addition, dissolution with ionic liquids resulted in coarse fibers with diameters of 75–110 μm, inferring poor keratin spinnability [21]. Formation of thick fibers could be because the obtained keratin was not linear in solution since ionic liquid mainly interrupted hydrogen bonds instead of disulfide bonds for crosslinking [33].
In this research, linear keratin with preserved backbones was obtained by breaking disulfide bonds connecting the keratin networks in wool. Strong surface electrical repulsion induced by addition of SDS led to controlled disentanglement and alignment of keratin molecules. The obtained fibers with diameters less than 20 μm verified good drawability of the keratin solution. Influence of additional alkaline treatments on mechanical properties of keratin fibers was also investigated.
Materials and methods
Materials
The wool fibers supplied from Brown Sheep Company (Mitchell, NE) were used because they were the major materials in wool-based municipal solid wastes. Sodium dodecyl sulfate (SDS, 99.0 %) was supplied by Hoefer Inc., San Francisco, CA, USA, and urea (99.0 %) was purchased from Oakwood Chemical Inc., West Columbia, SC. Coomassie Brilliant Blue R-250 (Proteomics Grade) was purchased from EMD Chemicals Inc., Gibbstown, NJ, USA. Other chemical reagents used in SDS-PAGE analysis, including LDS 9 sample buffer (4×), NuPAGE 20× MES running buffer and NuPAGE 4–12 % Bis–Tris gel, were from Invitrogen. Inc., Grand Island, NY. Cysteine (98.0 %) was purchased from Amresco LLC., Solon, OH. Calculation of concentrations took purity of chemicals into consideration.
The chemicals used in fiber spinning could be potentially green and sustainable, because they were either derived from renewable resources or could be reused. After extraction of wool keratin, urea could be easily recycled [34]. Cysteine, a standard amino acid with considerable reducibility, was an environmentally benign reductant, and could be commercially produced via fermentation [35]. SDS was synthesized from lauryl alcohol based on sulfur trioxide gas treatment. The lauryl alcohol was usually obtained after hydrolysis of vegetable oils, such as coconut oil or palm oil [36].
Controlled disintegration of disulfide crosslinks in wool keratin
The weight ratio of 8 M urea solution to wool of 17:1 was used to completely immerse wool, and the treatment temperature was 70 °C. Ten wt% of cysteine (based on wool fibers) was added into the solution. The pH was adjusted to 10.5 using 50 % NaOH solution. After 24 h, dispersion of wool in 8 M urea was centrifuged at 15000 rcf for 20 min to precipitate undissolved wool residue. The supernatant was adjusted to pH 4 using hydrochloric acid and sodium sulfate to precipitate dissolved keratin. The keratin precipitate was washed three times with distilled water under centrifugation of 15000 rcf for 20 min. The collected keratin was dried at 50 °C and pulverized using a mini Wiley mill with 20 mesh.
To study the influence of molecular weight on the mechanical properties of wool keratin, sodium hydroxide solutions of 0.05, 0.1, 0.25, and 0.5 % were used to treat the extracted wool keratin under 50 °C for 30 min at a liquor ratio of 10:1. The solutions were then neutralized using hydrochloric acid to pH 4 to precipitate the hydrolyzed keratin. The keratin precipitate was washed three times with distilled water under centrifugation of 15000 rcf for 20 min. The collected keratin was also dried at 50 °C and pulverized using a mini Wiley mill with 20 mesh.
SDS-PAGE
About 1 mg of extracted keratin from each treatment was dissolved in 100 μL NuPAGE® LDS Sample Buffer (1×), heated at 70 °C for 10 min and left standing at room temperature for 2 h. The solution was vortexed prior to loading. For each sample, 8 μL of solution was loaded into each slot of the gel. After electrophoresis, the gel was fixed in 65 % isopropyl ethanol/10 % acetic acid solution for 30 min, stained with 0.6 % Coomassie Brilliant Blue R-250 in 10 % acetic acid for 2 h at room temperature, and then destained using 10 % acetic acid until a clear background could be observed. The molecular weights of protein standard mixture ranged from 3 to 188 kDa.
Viscosity
Intrinsic viscosity of the extracted wool keratin and hydrolyzed keratin was determined according to ASTM standard D 2857 at a temperature of 25.0 ± 0.1 °C. An Ubbelohde capillary viscometer (size: 68b, capillary diameter: 0.55 mm) was used. The solvent was 0.3 M sodium carbonate buffer at pH 9.5.
Relative viscosity was conducted to investigate the effect of SDS addition on expansion and disentanglement of keratin molecules. About 16.7 % of extracted wool keratin and 5, 7.5, 8.75, 9.38, 10, 12.5, 15, 17.5, and 20 % of SDS (based on the weight of keratin) were mixed and dissolved in 0.3 M sodium carbonate-sodium bicarbonate buffer at pH 9.5. The solution was heated at 90 °C for 1 h. SDS solution under the same conditions without keratin was prepared as control. Apparent viscosity of the keratin solution and SDS solution was measured using a rotary rheometer (Brookfield, model R/S Plus, Middleboro, MA) with a CC25 DIN measuring system under the mode of CSR. About 25 g of solution was used for each test. The spindle and cup were immersed in a water bath at 90 °C throughout the test. The shear rate was set at 300 s−1 and the duration of each test was 1800 s. The relative viscosity was calculated by dividing the viscosity of solution by that of relevant SDS solution. Three specimens were tested for each condition.
Wet spinning of keratin fibers
Spinning dopes were prepared by adding 30 wt% of wool keratin and 10 wt% of SDS (based on weight of keratin) in 0.3 M sodium carbonate-sodium bicarbonate buffer (pH 9.5). The spinning dope was then allowed to age for 24 h at room temperature to enable full disentanglement of polypeptides. Before spinning, the solution was heated in water bath at 90 °C for 1 h. The fibers were then drawn manually into coagulation bath containing 10 % methanol and 10 % acetic acid. The fibers were washed in distilled water and dried under ambient conditions, then heated at 150 °C for 2 h and drawn manually twice and annealed at 120 °C for 1 h. The fibers were balanced in 21 °C and 65 % Relative humidity for 24 h prior to any test.
Morphological analysis
Scanning electron microscope (SEM, S3000 N, Hitachi Inc., Schaumburg, IL, USA) was employed to analyze the morphology of regenerated keratin fibers. Keratin fibers were sputter-coated with gold/palladium and observed at a voltage of 15 kV. Digital photos were also taken to compare the appearance of natural wool fibers and regenerated keratin fibers.
Tensile properties
Before tensile tests, fineness of the keratin fibers was measured in terms of denier, representing the weight of 9000 m of fibers in grams. Tensile properties of fibers in terms of breaking tenacity and breaking elongation were tested using an Instron tensile testing machine (Norwood, MA) according to ASTM standard D 3822. In the test, a gauge length of one inch and crosshead speed of 18 mm min−1 were used. For each condition, about 30 specimens were tested. Wet strength of keratin fibers was determined immediately after immersing the fibers in water at room temperature for 30 min.
Crystallinity analysis
X-ray diffraction study was carried out on raw wool fibers, extracted keratin powder and hydrolyzed keratin powder, and relevant keratin fibers. The data was collected on a Rigaku D/Max-B X-ray diffractometer with a CuKα radiation type and Bragg–Brentano parafocusing geometry, a diffracted beam monochromator, and a conventional copper target X-ray tube set (λ = 1.54 Å) to 40 kV and 30 mA at 26 °C. Diffraction intensities were recorded with 2θ ranging from 3° to 40° at a scan speed of 0.05 s−1. Crystallinity index (CI), indicating the relative crystallinity degree of fibers, was calculated using the following empirical eq. 1 [37].
where, CI is the crystallinity index; I9 is the maximum intensity of crystal lattice diffraction with 2θ at around 9°, and I14 is the minimum diffraction intensity with the 2θ at around 14°. In general, higher the CI value indicates higher crystallinity of the fiber sample.
Statistical analysis
All the data obtained were analyzed by the one-way analysis of variance with Scheffé test with a confidence interval of 95 %. A p value smaller than 0.05 indicated statistically significant difference. Standard deviations were shown by the error bars in figures, and the data in the figures labeled with different numbers or characters indicated significant differences among each of them.
Results and discussion
Controlled breakage of disulfide crosslinks
Dissolution process of wool included swelling of keratin macromolecules by urea and breakage of disulfide bonds by cysteine. Difficulty in dissolving wool stemmed mainly from their high crosslinking degrees, attributed to 10 % of cysteine in keratin [38–42]. In the high concentration urea solution, urea was inclined to disturb hydrogen bonds and weaken the hydrophobic interaction between polypeptides, and consequently exposed more polypeptides of wool keratin to the solvent. And thus, reductant could react with disulfide bonds within the peptide assembly. Comparing to mercaptoethanol, which was widely used in developing keratin-based biomaterials, the environmentally benign reductant cysteine could be biologically produced on a large scale.
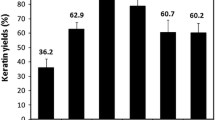
As seen from Fig. 1, lane 0 is the standard protein markers. Lane 1 is the raw wool, which did not dissolve in sample buffer attributed to the highly crosslinked network of keratin. Lane 2 is the extracted wool keratin with clear bands at 8, 15, 45 and 50 kDa. Lanes 3–6 are the wool keratin hydrolyzed under different alkaline concentrations. From lane 3 to lane 6, only two bands indicating molecular weights of around 8 kDa existed at the bottom. Other than that, smear spread from top to bottom, indicating random cleavage of polypeptides in the keratin under different alkaline conditions. However, the smear of keratin extracted in 0.05 % alkaline was slightly darker than those from the other three concentrations. The mild extraction condition used for keratin in lane 2 did not seriously destroy the backbones of wool keratin, and led to yield of approximate 63 %. Considering the good extraction yield and preservation of molecular structures of keratin, it could be suggested that the extraction method may be feasible for large-scale production.

SDS-PAGE of extracted wool keratin indicating that the raw wool was not soluble, the extracted wool keratin had major bands at 45 and 50 kDa, while the hydrolyzed wool keratin did not show detectable bands at molecular weight higher than 10 kDa. Lane 0 is the standard protein marker, lane 1 is raw wool, lane 2 is the keratin extracted using 8 M urea at pH 9.5 for 24 h, lane 3, 4, 5, 6 are the wool keratin hydrolyzed in sodium hydroxide solutions with concentrations of 0.05, 0.1, 0.25 and 0.5 %, respectively
Intrinsic viscosity
Figure 2 indicates that intrinsic viscosity of the wool keratin increased as extraction conditions weakened. It could be observed that the intrinsic viscosity decreased as the concentration of sodium hydroxide increased, suggesting gradual weakening of intra- and inter-molecular interactions among keratin macromolecules. Cleavage of more keratin polypeptides in NaOH solution with higher concentrations created more short polypeptides with polar end amine or carboxyl groups, and thus weakened molecular interaction among keratin polypeptides.

Intrinsic viscosity of wool keratin increased as the treatment conditions weakened. The first 4 bars represented wool keratin hydrolyzed in sodium hydroxide solutions with concentrations of 0.5, 0.25, 0.1 and 0.05 % at 50 °C for 30 min with a liquor ratio of 10:1. The last bar indicated with 0 represents wool keratin obtained from the urea/cysteine solvent system
Relative viscosity
SDS endowed keratin solution with spinnability by enhancing disentanglement and alignment of macromolecules. Polymers usually entangled with each other in solution. Spinnability indicated the capability of polymer solution to elongate irreversibly under stretching. Optimal fibers could be obtained if the entangled polymers were unraveled and became aligned, while a small portion of polymers were left hooked to each other.
Figure 3 demonstrates that the viscosity of keratin dispersion or solution first increased and then decreased as concentration of SDS increased, achieved maximum viscosity as SDS concentration reached 10 %. Further increase in SDS concentration decreased viscosity to around one sixth of the peak value. The SDS in the keratin dispersion determined the entanglement of macromolecules in the aqueous environments. With low amount of SDS, the insoluble keratin in water agglomerated instead of dissolved. The low increase in viscosity of solution with SDS concentrations from 5 to 9 % was mainly attributed to the low interaction among water molecules. The sharp increase in viscosity from 8.75 to 10 % indicated enhanced interaction among protein molecules, confirming the successful disentanglement of keratin with high SDS concentration. However, further addition of SDS from 10 wt% rapidly decreased viscosity. It could be inferred that the remained entanglement among keratin molecules may be completely dissociated by excess SDS, while electrical repulsion among molecules was increasingly enhanced, since more SDS could join existing micelles on the surface of molecules. Breaking of entanglement and enhancement in molecular repulsion significantly weakened molecular interaction as contacting area of liberated molecules was further reduced. At this concentration, intensified interaction among SDS micelles on keratin molecules may dominate the decrease of viscosity. In fiber spinning, low SDS concentration resulted in poor alignment of molecules and thus unsatisfied drawability of polymers, while high SDS concentration that led to weak interaction among molecules could result in poor physical properties of fibers. Therefore, the SDS of 10 wt %, at which concentration keratin molecules showed strongest interaction and appropriate disentanglement was selected for fiber spinning.

Influence of concentration of SDS on relative viscosity of keratin spinning dope. The viscosity increased till the SDS concentration reached 10 % and then decreased
Morphology in micro- and macro- scales
As shown in Fig. 4a, the regenerated wool fiber with uniform diameter of around 20 μm had crenulations along the longitudinal axis, indicating the featured sheath-core structures of wet spun fibers. In Fig. 4b, a fiber with diameter of around 30 μm was intentionally selected to demonstrate the morphological details. The cross-section of the regenerated fiber was irregular and indented, suggesting uneven shrinking of fibers during solidification of fibers in the coagulation bath. In addition, the smooth cross-section indicated brittle fracture of the fiber during breakage. The small diameters of the fibers proved good spinnability of wool keratin in the solution.

SEM images of a longitudinal and b cross-sectional views of regenerated keratin fibers. The fibers with deep grooves along the axis indicated typical morphologies of wet spun fibers and the smooth cross-section indicated brittle fracture. (spinning conditions: 30 % protein in 0.3 M carbonate-bicarbonate buffer, 10 % SDS based on the weight of keratin, coagulation bath with 10 % methanol and 10 % acetic acid)
Figure 5 shows that the raw wool fibers on the left have significantly bigger diameters than the regenerated wool keratin fibers on the right. Therefore, the regenerated keratin fibers with smaller diameters might have softer hand than the raw wool fibers. The length of regenerated wool keratin fibers could be longer than two meters, much longer than the raw wool fibers. Therefore, continuous filaments and multifilament yarns might be directly obtained in pilot-scale production.

Digital photos of raw wool fibers on the left and regenerated wool keratin fibers on the right, indicating smaller diameters of the regenerated keratin fibers than natural wool fibers
Tensile strength
Mechanical properties of the regenerated keratin fibers at dry and wet states are shown in Fig. 6. The dry tensile strength of fibers from wool keratin with no alkaline treatment was approximate 0.88 g/denier, which reached the lower limit of tensile strength of natural wool fibers, and was significantly higher than 0.79, 0.68, 0.52, 0.49 g/denier, that of fibers from wool keratin hydrolyzed in sodium hydroxide solutions with increasing concentrations. The wet tensile strength of fibers of around 0.26 g/denier was not significantly different from each other, and was still much higher than that of regenerated soyprotein fibers, which was around 0.17 ± 0.03 g/denier [43]. The better wet strength of keratin fibers could be attributed to the remained crosslinks among the molecules. The dry elongation of regenerated keratin fibers of about 10 % and the wet elongation of about 25 % were much lower than that of other regenerated protein fibers. Comparing to other reported keratin based materials (Table 1), such as crosslinked films, blended films, and blended fibers, the regenerated keratin fibers showed significantly higher tensile properties and better extensibility. The results also could be due to the better alignment of macromolecules in the keratin fibers obtained in this work. Fig. 7 demonstrated typical stress–strain curves of dry and wet fibers from 0.05 % NaOH treated wool keratin. The elongation of fibers under wet state was much larger than under dry state.

Effect of different alkaline treatments on the tensile properties, including dry and wet tensile strength of regenerated keratin fibers. From left to right, wet spun fibers from un-hydrolyzed wool keratin, wool keratin hydrolyzed in sodium hydroxide solutions with concentrations of 0.05, 0.1, 0.25 and 0.5 % at 50 °C for 30 min with a liquor ratio of 10:1. Different letters for each bar indicated significant differences among them

Typical stress strain curves of fibers from 0.05 % NaOH treated wool keratin under dry and wet states
The regenerated wool fibers could be blended with wool, cotton or synthetic fibers to make yarns and other industrial products. Furthermore, mechanical properties of the fibers could be improved to meet the industrial standards via many widely used approaches, such as chemical crosslinking and nanoparticle filling in the future.
Figure 8 shows that the intrinsic viscosity and dry state tensile strength have linear relation with a coefficient of 20.398 and r square of 0.94. This correlation indicated that fibers composed of longer polypeptides showed higher tensile strength. Higher intrinsic viscosity indicated longer macromolecules in the spinning solution which led to stronger interactions and as a result higher strength.

Simulated linear relation between dry-state tensile strength of regenerated keratin fibers and intrinsic viscosity of wool keratin after alkaline treatments in sodium hydroxide solutions with concentrations of 0.05, 0.1, 0.25 and 0.5 % at 50 °C for 30 min with a liquor ratio of 10:1
Crystalline structure
As shown in Fig. 9, the decreasing trend of CI was also in accordance with the difference in tensile properties from raw wool to regenerated fibers after hydrolysis in sodium hydroxide solutions with concentration of 0.05, 0.1, 0.25 and 0.5 %, respectively. The raw wool fibers had the highest crystallinity index of around 55 %, which was consistent with other reported results [49]. The keratin powder showed even lower CI value, indicating even lower crystallinity. It was suggested that the precipitated keratin in either form of powders or fibers could not completely reconstruct the tight arrangement of polypeptides existed in the raw wool. It has been proved in literatures that aqueous organic solvent induced decline in alpha-helix and increase in beta-sheet [50]. Being precipitated in alcohol, much tighter molecular structures, probably beta-sheets, could be partially rebuilt in the protein fibers, and resulted in better mechanical performances.

Crystallinity indices of raw wool fibers and regenerated wool keratin fibers and wool powders after hydrolysis in sodium hydroxide solutions with concentrations of 0.05, 0.1, 0.25 and 0.5 % at 50 °C for 30 min with a liquor ratio of 10:1. The keratin powder showed even lower CI value, indicating even lower crystallinity
X-ray diffraction spectrograms of the raw wool and regenerated keratin fibers are shown in Fig. 10. Since all the regenerated keratin fibers had similar patterns, only the pattern of regenerated fiber with no alkaline treatment was selected. Both of the two spectrograms were typical diffraction pattern of α-keratins with a prominent 2θ peak at 20.2° and a minor peak at 9°, indicating the crystalline spacing of 4.4 and 9.8 Å, respectively [51]. Wider peaks implied smaller crystalline structures in the fibers. It could be observed that raw wool with larger peak at 9° than 20° had more larger crystals, while the regenerated keratin fibers with larger peak at 20° than 9° had more smaller crystals.

Typical X-Ray Diffraction spectrograms with peaks at 9° and 20° showing typical alpha-helix configurations of raw wool fibers and regenerated keratin fibers without extra alkaline hydrolysis
Conclusion
Effective dissolution of wool keratin was achieved via cleavage of disulfide bonds while preserving the backbones, and verified through successful spinning of regenerated keratin fibers with diameters as small as 20 μm. The mechanical properties of the regenerated keratin fibers were in agreement with their differences in molecular weight, intrinsic viscosity and crystallinity index. Successful fiber spinning indicated the possibility of successful development of macromolecules in films, sponges and many other forms. Broad impact of the dissolution method could be resulted in exploring applications of other largely available highly crosslinked proteins, such as sorghum proteins and chicken feathers from biofuel and agricultural industries, which could be developed into useful industrial products, such as films, sponges and fibers.
References
Shishoo R (2012) Technological advances and future challenges. The global textile and clothing industry. Elsevier, Amsterdam, pp 8–28
Rauschendorfer LM (2013), Global Development of Fiber Production. http://www.chemistryviews.org/details/news/4723361/Global_Development_of_Fiber_Production.html. Accessed 16 June, 2013
Johnson J, MacDonald S, Meyer L, Norrington B and Skelly C (2014), The World and United States Cotton Outlook,http://www.usda.gov/oce/forum/2014_Speeches/Cotton.pdf. Accessed 2 May, 2014
Chen HL, Burns LD (2006) Environmental analysis of textile products. Cloth Textiles Res J 24:248–261
U.S. Environmental Protection Agency (2013). http://www.epa.gov/osw/conserve/materials/textiles.htm. Accessed 26 Feburary, 2014
Food and Agricultural Organizations of the United Nations (2013). http://faostat.fao.org/site/569/DesktopDefault.aspx?PageID=569#ancor. Accessed 26 Feburary, 2014
Poole AJ, Church JS, Huson MG (2008) Environmentally Sustainable Fibers from Regenerated Protein. Biomacromolecules 10:1–8
Wen G, Naik R, Cookson P, Smith S, Liu X, Wang X (2010) Wool powders used as sorbents to remove Co2+ ions from aqueous solution. Powder Technol 197:235–240
Rajkhowa R, Zhou Q, Tsuzuki T, Morton DA, Wang X (2012) Ultrafine wool powders and their bulk properties. Powder Technol 224:183–188
Yang Y, Reddy N (2013) Potential of using plant proteins and chicken feathers for cotton warp sizing. Cellulose 20(4):2163–2174
Huda S, Yang Y (2009) Feather Fiber Reinforced Light-Weight Composites with Good Acoustic Properties. J Polym Environ 17:131–142
Hu C, Reddy N, Yan K, Yang Y (2011) Acetylation of chicken feathers for thermoplastic applications. J Agri Food Chem 59:10517–10523
Sun P, Liu ZT, Liu ZW (2009) Chemically Modified Chicken Feather as Sorbent for Removing Toxic Chromium (VI) Ions. Ind Eng Chem Res 48:6882–6889
Yang Y, Reddy N (2013) Potential of using plant proteins and chicken feathers for cotton warp sizing. Cellulose 20:2163–2174
Harris M, Brown AE (1947) Natural and Synthetic Protein Fibers. Text Res J 17:323–330
Evans RL, Shore B (1948) U.S. Patent. 2 434 688, Regenerated Keratin
Wormell RL, Happey F (1949) Regenerated Keratin Fibres. Nature 163:18
USDA “Value Added Products from Feather Fiber” $175 K (2004–2006), http://www.eng.auburn.edu/users/royalb/EXPERIENCE.htm (Accessed March 5, 2014)
USDA awards grant $500,000 to develop bio-based products http://www.udel.edu/PR/UDaily/2005/mar/rwool080405.html (Accessed March 5, 2014)
Idris A, Vijayaraghavan R, Rana UA, Fredericks D, Patti AF, MacFarlane DR (2013) Dissolution of feather keratin in ionic liquids. Green Chem 15:525–534
Fan X (2008) Value-added products from chicken feather fibers and protein. Doctoral Thesis. PhD Dissertation, Auburn University, Auburn
Tonin C, Aluigi A, Vineis C, Varesano A, Montarsolo A, Ferrero F (2007) Thermal and structural characterization of poly (ethylene-oxide)/keratin blend films. J Therm Anal Calorim 89:601–608
Aluigi A, Vineis C, Ceria A, Tonin C (2008) Composite biomaterials from fibre wastes: Characterization of wool–cellulose acetate blends. Composites Part A Appl S 39:126–132
Hameed N, Guo Q (2010) Blend films of natural wool and cellulose prepared from an ionic liquid. Cellulose 17:803–813
Iridag Y, Kazanci M (2006) Preparation and characterization of Bombyx mori silk fibroin and wool keratin. J Appl Polym Sci 100:4260–4264
Vasconcelos A, Freddi G, Cavaco-Paulo A (2008) Biodegradable materials based on silk fibroin and keratin. Biomacromolecules 9:1299–1305
Yuan J, Xing ZC, Park SW, Geng J, Kang IK, Shen J, Meng W, Shim KJ, Han IS, Kim JC (2009) Fabrication of PHBV/keratin composite nanofibrous mats for biomedical applications. Macromol Res 17:850–855
Barone JR, Schmidt WF, Liebner CF (2005) Thermally processed keratin films. J Appl Polym Sci 97:1644–1651
Barone JR, Schmidt WF, Gregoire N (2006) Extrusion of feather keratin. J Appl Polym Sci 100:1432–1442
Desnuelle P (1953) In: Neurath H, Bailey K (eds) The Proteins, vol I, Part A. Academic Press, New York, p 87
Jia QX, Xiong ZJ, Shi CM, Zhang LQ, Wang XN (2012) Preparation and properties of polyamide 6 fibers prepared by the gel spinning method. J Appl Polym Sci 124:5165–5171
Tsen C (1969) Effects of oxidizing and reducing agents on changes of flour proteins during dough mixing. Cereal Chem 46:435–442
Ghosh S, Banerjee A (2001) A Multitechnique Approach in Protein/Surfactant Interaction Study: Physicochemical Aspects of Sodium Dodecyl Sulfate in the Presence of Trypsin in Aqueous Medium. Biomacromolecules 3:9–16
Behrendt J, Arevalo E, Gulyas H, Niederste-Hollenberg J, Niemiec A, Zhou J, Otterpohl R (2002) Production of value added products from separately collected urine. Water Sci Technol 46:341–346
Wada M, Takagi H (2006) Metabolic pathways and biotechnological production of l-cysteine. Appl Microbiol Biotechnol 73:48–54
Gallezot P (2007) Process options for converting renewable feedstocks to bioproducts. Green Chem 9:295–302
Long JJ, Cui CL, Wang L, Xu HM, Yu ZJ, Bi XP (2013) Effect of treatment pressure on wool fiber in supercritical carbon dioxide fluid. J Clean Prod 43:52–58
Elsworth FF, Phillips H (1941) The action of sulphites on the cysteine disulphide linkages in wool: The influence of temperature, time and concentration on the reaction. Biochem J 35:135–143
Xu H, Cai S, Xu L, Yang Y (2014) Water-stable three-dimensional ultrafine fibrous scaffolds from keratin for cartilage tissue engineering. Langmuir 30:8461–8470
Xu H, Yang Y (2014) Controlled De-Cross-Linking and Disentanglement of Feather Keratin for Fiber Preparation via a Novel Process. ACS Sustain Chem Eng 2:1404–1410
Xu H, Cai S, Sellers A, Yang Y (2014) Electrospun ultrafine fibrous wheat glutenin scaffolds with three-dimensionally random organization and water stability for soft tissue engineering. J Biotechnol 184:179–186
Xu H, Cai S, Sellers A, Yang Y (2014) Intrinsically water-stable electrospun three-dimensional ultrafine fibrous soy protein scaffolds for soft tissue engineering using adipose derived mesenchymal stem cells. RSC Adv 4:15451–15457
Reddy N, Li Y, Yang Y (2009) Alkali-catalyzed low temperature wet crosslinking of plant proteins using carboxylic acids. Biotechnol Prog 25:139–146
Tanabe T, Okitsu N, Tachibana A, Yamauchi K (2002) Preparation and characterization of keratin–chitosan composite film. Biomaterials 23:817–825
Tanabe T, Okitsu N, Yamauchi K (2004) Fabrication and characterization of chemically crosslinked keratin films. Mater Sci Eng, C 24:441–446
Katoh K, Shibayama M, Tanabe T, Yamauchi K (2004) Preparation and physicochemical properties of compression-molded keratin films. Biomaterials 25:2265–2272
Vasconcelos A, Freddi G, Cavaco-Paulo A (2008) Biodegradable Materials Based on Silk Fibroin and Keratin. Biomacromolecules 9:1299–1305
Katoh K, Shibayama M, Tanabe T, Yamauchi K (2004) Preparation and properties of keratin–poly(vinyl alcohol) blend fiber. J Appl Polym Sci 91:756–762
Hou X, Xu H, Shi Z, Ge M, Chen L, Cao X, Yang Y (2014) Hydrothermal pretreatment for the preparation of wool powders. J Appl Polym Sci. doi:10.1002/app.40173
Tsukada M, Freddi G, Monti P, Bertoluzza A, Kasai N (1995) Structure and molecular conformation of tussah silk fibroin films: Effect of methanol. J Polym Sci, Part B: Polym Phys 33:1995–2001
Xu W, Cui W, Li W, Guo W (2004) Development and characterizations of super-fine wool powder. Powder Technol 140:136–140
Acknowledgements
This research was financially supported by the Agricultural Research Division at the University of Nebraska-Lincoln, USDA Hatch Act, Multistate Research Project S-1054 (NEB 37-037) and Nebraska Environmental Trust (13-142). The authors thank the AATCC student research grant for Zhuanzhuan Ma. The authors also thank Dr. Han Chen for his help in SEM.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, H., Ma, Z. & Yang, Y. Dissolution and regeneration of wool via controlled disintegration and disentanglement of highly crosslinked keratin. J Mater Sci 49, 7513–7521 (2014). https://doi.org/10.1007/s10853-014-8457-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8457-z