
Abstract
Tungsten oxide (WO3) nano-ribbons (NRs) were obtained by annealing tungstite (WO3·H2O) NRs. The latter was synthesized below room temperature using a simple, environmentally benign, and low cost aging treatment of precursors made by adding hydrochloric acid to diluted sodium tungstate solutions (Na2WO4·2H2O). WO3 generates significant interests and is being used in a growing variety of applications. It is therefore important to identify suitable methods of production and better understand its properties. The phase transformation was observed to be initiated between 200 and 300 °C, and the crystallographic structure of the NRs changed from orthorhombic WO3·H2O to monoclinic WO3. It was rigorously studied by annealing a series of samples ex situ in ambient air up to 800 °C and characterizing them afterward. A temperature-dependent Raman spectroscopy study was performed on tungstite NRs between minus 180 and 700 °C. Also, in situ heating experiments in the transmission electron microscope allowed for the direct observation of the phase transformation. Powder X-ray diffraction, electron diffraction, electron energy-loss spectroscopy, and X-ray photoelectron spectroscopy were employed to characterize precisely this transformation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tungsten oxide (WO3) is an n-type semiconductor with a widely tunable band gap, varying from Eg ≈ 2.5–2.8 eV at room temperature to ≈1.7 eV at 800 °C [1]. WO3 generates significant interests and is being used in a growing variety of applications. It is therefore important to identify suitable methods of production and better understand its properties. WO3 thin films and nanoparticles [2] were investigated for use in different applications: for example, in microelectronics and optoelectronics [3, 4], dye-sensitized solar cells [5, 6], solid-state electrochromic cells [7], colloidal quantum dot light emitting diodes [8], photocatalysis [9], photoelectrocatalysis [10, 11], water splitting photocatalyst as visible-light-absorbing semiconductor [12–20], conversion of cellulose to ethylene glycol [21], methanol oxidation catalyst [22], and alcohol dehydration [23]. WO3 has also been tested for ultra-fast bleaching-coloring performance for the development of windows [24] in energy-efficient buildings, flat-panel displays, optical memory, and writing-reading-erasing devices. All applications are strongly dependent on the structure of WO3 because its properties are directly linked to it. Though, there exist some publications reporting on the WO3 phase transitions, they were carried for materials having very different morphologies and also using different experimental methods and conditions [25–28].
This article reports on the phase transformation of tungstite (WO3·H2O) nano-ribbons (NRs, TNRs) to WO3 NRs (WNRs) by annealing. A good understanding of this mechanism is important because the crystal structure of WO3 is very prone to temperature [29], stoichiometry, and dimensions, directly affecting its physical properties, such as its electrical conductivity [30]. This transformation was rigorously studied by annealing a series of tungstite samples ex situ in ambient air up to 800 °C and characterizing them afterward. In situ heating experiments in the transmission electron microscope (TEM) allowed for the direct observation of this phase transformation. Because this phase transformation is actually the dehydration of tungstite NRs, a definite advantage of in situ experiments (in high vacuum) is the ability to observe what happens in real time and therefore avoid water (from the ambient atmosphere) to absorb on these materials. Also, in situ heating experiments in the TEM offer unsurpassed control of the heating cycles and high lateral resolution, and provide additional benefits such as the ability to record selected area electron diffraction (SAED) patterns and electron energy-loss spectra (EELS) in real time [31, 32]. A temperature-dependent Raman spectroscopy study was performed on tungstite NRs between minus 180 and 700 °C, during which the structural phase transformation was detected because of the variations in the lattice vibrations. X-ray diffraction (XRD), SAED, EELS, and x-ray photoelectron spectroscopy (XPS) were all employed to characterize precisely this phase transformation in a large temperature window.
Experimental details
In the first step, the tungstite (WO3·H2O) NRs were synthesized using the acid precipitation method [33, 34]. Around 30–50 mL of 6 N hydrochloric acid (HCl) was added drop wise to a 100 mL 15 mM sodium tungstate solution (Na2WO4·2H2O), while this solution was kept at 5–10 °C and under constant stirring. The precipitation started to take place when about 30 mL of HCl had been added. However, in order to increase the yield, up to 50 mL of HCl was added. Ultrapure 18 MΩ Millipore® deionized water was used for the preparation of all solutions. The solutions were centrifuged and washed to reach pH 6 and the products added to abundant aqueous solutions for crystallization and aging for durations of 24–48 h at room temperature (RT) under constant stirring.
The samples were examined using a field-emission SEM (FESEM, JEOL JSM-7500F) and a high resolution TEM (HRTEM, JEOL JEM-2200FS, operated at 200 kV) both equipped with X-ray energy-dispersive spectrometers (XEDS), used to determine the elemental composition of the samples. The TEM is equipped with an in-column energy filter and was used to record EEL spectra and energy filtered images. In situ experiments were carried out using a commercial specimen holder (Aduro by Protochips). Pictures showing the experimental set up are displayed in Fig. 1. A drop of a suspension of tungstite NRs (TNRs) dispersed in ethanol was placed and allowed to dry on interdigitated heating devices (Fig. 1b). One calibrated device was used for each experiment. The ramp rate was set at 10 °C.min−1 in all cases, just like all other ramp rates in this study (matching the in situ Raman and ex situ annealing). The phase transformation was monitored using imaging, SAED, and EELS. The TNRs and WNRs being sensitive to the electron beam [35, 36], exposure times, and illumination—to the electron beam—were kept at a minimum. EEL spectra and SAED patterns were recorded from new NRs each time. During ramping and heating, the electron beam was kept off the sample (i.e., the beam valve was closed). Therefore, the heating is assumed to be solely caused by the heating applied using the in situ heating specimen holder.

Pictures showing the experimental set up for the in situ TEM heating experiments. a Power supply and specimen holder. b The actual device and SEM image showing the architecture of this specific interdigitated heating device. The TNRs suspended in ethanol were dispersed onto this device, allowed to dry, and characterized (through the carbon support film in the holes)
Thermo gravimetric analysis (TGA, SDTA 851, Mettler Toledo) plots were recorded at 10 °C.min−1 to determine the proper heat treatment temperatures to be used in subsequent experiments.
Temperature-dependent Raman scattering experiments were performed on dried TNRs (they were dried at 60 °C for 10–12 h) in vacuum. The spectrometer used was a Horiba Jobin–Yvon T64000 micro-Raman system, with an excitation of 532 nm wavelength from a diode LASER. The energy of the LASER beam was kept low (0.2 mW) to avoid any damage to the samples. The temperature was varied from minus 185 to 700 °C, with a ramp rate of 10 °C.min−1.
The XPS measurements were performed using a PHI 5500 spectrometer operated using a monochromated Al Kα radiation (1,487 eV) and an electron emission angle of 45°. In order to compare the XPS data with the in situ TEM and Raman spectroscopy observations, XPS was performed on a set of samples that were heat-treated ex situ at different temperatures (from 300 to 800 °C) in a tube furnace in ambient air (Lindberg/Blue M Mini-MiteTM).
The current versus voltage (I–V) and current transient characteristics of these materials were recorded using interdigitated Pt/PtO electrode devices onto which drops of the suspensions (the materials were dispersed in ethanol) were placed and allowed to dry. The electrode separation distance was 20 µm with an electrode finger length of 1 mm. A picture of the actual device is displayed in Fig. 11a as inset. A high internal resistance Keithley 2401 electrometer was used to record the signals. UV/Visible (UV/Vis) absorption spectra in aqueous solutions were recorded using Varian Cary 500 spectrophotometer.
Results and discussion
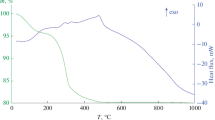
Figure 2 shows the TGA plot recorded from the TNRs and revealed two mass loss steps. The first loss (I) occurred below 100 °C and corresponded to the evaporation of absorbed water; it confirms the sensitivity of this material to water adsorption. The second loss (II) occurred between 170 and 300 °C and was due to the elimination of inter-structural water molecules [34].

TGA plot recorded from TNRs (ramp was 10 °C.min−1)
Here, we report on the results obtained from a set of eight samples, with conditions listed in Table 1. They were each given a sample ID.
SEM images showing as-synthesized and heat-treated NRs (in the furnace) at different temperatures are displayed in Fig. 3a–g. XEDS recorded in the SEM and the TEM showed that only O and W elements were present in all the samples (note that no organic compounds were used in the synthesis). The TNRs were less than 1-µm long (average length of 600 nm [34]), 150–550 nm wide, and about 20 nm thick. The heat treatment in air (i.e., oxygen is present in the medium of the furnace) resulted in significant changes in the morphology of the NRs for temperatures in excess of 600 °C. Images shown in Fig. 3e–g confirm that significant sintering occurred at temperatures higher than 600 °C. Inter-structural water molecules were removed leading to the phase transformation from orthorhombic tungstite to monoclinic (a more compact structure) tungsten oxide. It causes tension and/or cracks, even though slow ramping rates were used. The NRs preserved their sharp edges below 500 °C (e.g., sample W500), but the morphology was not preserved for temperatures higher than 600 °C (samples W600, W700, and W800) with a change from a 2D to a 3D structure. A similar sintering phenomenon was reported for tungsten oxide nanodisks which turned to 3D particles when the temperature was increased from 500 to 700 °C [37]. The goal is to preserve this 2D nanoribbon morphology in order to maintain the large active surface area for their use as catalysts. The consequence of this sintering is also apparent in the photograph shown in Fig. 3i; it was taken 24 h after six different samples that were subjected to different heat treatment temperatures (and dispersed in water) were ultrasonicated for 5 min. The NRs that were treated up to 500 °C took several days to settle to the bottom of the containers, while the NRs that were treated above 500 °C settled in only few hours which is a direct correlation of their sintering. The nanoribbon morphology brings a very clear advantage for catalysis and photocatalysis applications, because they can remain suspended for very long time without any external force.

SEM images showing TNRs obtained by aging a WO3 NRs obtained by sintering in air at temperature b T = 300 °C (W300), c 400 °C (W400), d 500 °C (W500), e 600 °C (W600), f 700 °C (W700), and g 800 °C (W800). h XRD recorded from different samples before and after heat treatments, and i a photograph taken 24 h after ultrasonication for 5 min of six suspensions of TNRs treated at various temperatures and dispersed in water
X-ray diffractograms recorded from the TNRs showed it to have the orthorhombic Pmnb structure of tungstite with lattice parameters a = 0.524, b = 1.070, and c = 0.512 nm, which was in good agreement with published data (WO3·H2O JCPDS No. 43-0679 and the American Mineralogist Crystal Structure Database, AMCSD 0005199 [38] ). This orthorhombic structure consists of distorted octahedral units of tungsten atoms coordinated with five oxygen atoms and a water molecule where the octahedra share four vertices in the equatorial plane forming the sheet structure. The sheets are held together by weak (van der Waals and/or hydrogen) bonding between the water molecules and the oxygen atoms in the axial position (z direction) in the adjacent layers. The bond lengths between tungsten and oxygen are 0.169 nm (W = O), two 0.183 nm (W–O), two 0.193 nm (W–O), and 0.234 nm (axial, water W–OH2) [38].
XRD showed that annealings to temperatures above 300 °C resulted in a phase transformation to the monoclinic P21/n (14) structure with lattice parameters a = 0.729, b = 0.754, and c = 0.768 nm, which was in good agreement with published data (WO3 JCPDS No. 43–1035). In this monoclinic structure, W–O bonds make zigzag chains in three directions with W–O–W angles of 158° and O–W–O angles of 166°, while in the x direction, the bonds show equal length, while in the y and z directions, they are alternately long and short [39]. These two structures are illustrated in the schematics of Fig. 4. The WO6-octahedra are arranged in corner sharing (e.g., ReO3-type structure) in the (xy) plane and held together by oxygen (W–O–W) bonds, perpendicular to the < 001 > axis. It leads to the formation of tunnels between the octahedras where small ions can be placed. It may allow for ionic transport and intercalation in these structures.

Schematic of the orthorhombic unit cell of tungstite which converts to the monoclinic tungsten oxide unit cell by dehydration at elevated temperatures (≥300 °C)
The orthorhombic TNRs transform relatively fast to monoclinic NRs at T ≥ 300 °C (the transformation is actually initiated between 200 °C and 300 °C as shown later in the Raman spectra), by the removal of inter-structural water molecules. The first step being the protonation [40] of the tungstate ions upon solution acidification to form white solid precipitates
The second step is the hydration of the [WO2(OH)2] tetrahedral molecules and dimerization via O-bridging to form crystalline [WO(OH)3(H2O)]2(µ-O) containing octahedral W-centers [40]:
Details of these mechanisms are shown elsewhere [34]. And finally, the tungstite transforms to tungsten oxide by dehydration:
The application of temperature on tungstite leads to a phase transition from orthorhombic tungstite to monoclinic tungsten oxide. This transition requires the removal of the inter-structural water molecules which can cause stress to the thin ribbons (See Fig. 3b, c, d). The ribbon morphology was preserved at temperatures below 600 °C but for higher temperatures, the materials turned to 3D particles, which present drawbacks for applications (i.e., reduced surface area and impaired floatation). The schematic of this dehydration process is shown in Fig. 4.
The removal of inter-structural water molecules and W=O double bonds (which is labeled as O(2) in Fig. 4) allows for the layers to connect through O atoms to stack together and turn to a more compact structure. The theoretical densities of orthorhombic tungstite and monoclinic tungsten oxide are 5.78 gr.cm−3 and 7.28 gr.cm−3, respectively [38 and JCPDS No. 43-1035].
The results of the temperature-dependent Raman spectroscopy study conducted on sample WRT1 (TNRs) are displayed in the two plots of Fig. 5a, b, where the spectra recorded from 0 to minus 185 °C and from room temperature (RT) to 700 °C are shown in (a) and (b), respectively. The Raman peaks in the spectral range of 400–1,000 cm−1 are associated with WO6 stretching modes. There are two intense Raman peaks at about 635 (broad ω1) and 943 (sharp ω2) cm−1. The broad peak ω1 and the sharp peak ω2 are associated with the stretching mode of the W6+-O(2) and W6+-O(3), respectively (see Figs. 4, 5). The bond length of W6+-O(2) is larger than that of W6+-O(3), hence ω1 appears at lower frequency than ω2. These two peaks are characteristic peaks of orthorhombic WO3·H2O. A decrease in temperature resulted in all the peaks to show a blue shift. Moreover, significant changes were observed in the peaks appearing in the low frequency spectral region. The intensity of the peak at 41 cm−1 increases drastically with a decrease in the temperature. It may be pointed out that except for the change in peak position, no extra Raman peak was observed in the low temperature region. This indicates that tungstite does not undergo any structural phase transition at low temperature.

Temperature-dependent Raman spectroscopy measurements performed on WRT1 sample a in the range of minus 185 °C to RT and b in the range of RT to 700 °C. c Raman spectra recorded at 250 °C and 300 °C showing the presence of the two phases (orthorhombic and monoclinic)
No changes occurred when the temperature was increased up to 100 °C. At 200 °C, few changes in the spectra were observed: a new mode appeared at about 845 cm−1, the intensity of one of the stretching modes (ω2 = 943 cm−1) decreased significantly, and the other stretching mode (ω1 = 645 cm−1) became asymmetrically broader (shifted to higher wave number). These modes were fitted with four Lorentzian peaks as shown in Fig. 5c. Note that the broad peak has been fitted with two peaks, the peak fitted at lower wavenumber represents ω1 vibration of orthorhombic phase, whereas the peak fitted at higher wavenumber (ω3 = 700 cm−1) along with the peak at (ω4 = 810 cm−1) representing the characteristic stretching vibrational modes of monoclinic WO3. This observation indicates that at 200 °C both orthorhombic and monoclinic phases coexisted. As the temperature was further increased to 250 °C, all Raman peaks belonging to the orthorhombic phase disappeared. At this temperature, the stretching vibrations (ω3 and ω4) related to the monoclinic phase became more intense, and changes were also observed in the low frequency region of the spectra. For clarity, the Raman spectra for the mixed phase and the pure monoclinic phase are compared in Fig. 5c.
The core level XPS spectra recorded from samples TNRs, W300, W500, and W700 samples are presented in Fig. 6. The spectra for all samples recorded in the region of W4f showed W4f 7/2 and W4f 5/2 doublet at the binding energies of 35.6 and 37.7 eV (with peak separation of 2.1 eV or 17,000 cm−1 and the intensity ratio of 3:4) and are associated with oxidation state W6+ ions [41] (see Fig. 6a). The O1 s spectrum of each sample was fitted with three peaks (see Fig. 6b). The main contribution to the O1 s spectra at the lowest binding energy originates from the W–O bond. The second peak at about 532.5 eV represents the –OH bond, corresponding to the absorbed or inter-structural water molecules inside the tungsten oxide or the tungstite structure. The highest intensity for this peak was observed in the TNRs sample because of the inter-structural water molecules (see Figs. 4, 6b). The intensity of this second peak was observed to increase for samples that were heat treated to 300, 500, and 700 °C, which shows increasing sensitivity to the adsorption of water on their surface. The third small peak in the O1 s spectra can be assigned to the O–C bonds representing adsorbed contaminations, commonly detected in XPS.

XPS O1 s and W4f core level spectra recorded from TNRs, W300, W500, and W700 samples
One TEM image and a corresponding SAED pattern recorded at RT from as-synthesized TNRs are shown in Fig. 7a, g. This diffraction pattern (DP) was indexed to the orthorhombic Pmnb structure of tungstite, which is in good agreement with XRD. The images shown in (b–f) were recorded during the in situ heating experiments in the TEM at 300, 500, 600, 700, and 800 °C, respectively. There were no signs of sintering between NRs unlike when heating was performed in the presence of oxygen (i.e., in the furnace; see Fig. 3) because of their intentionally reduced concentration.

a TEM image recorded from TNRs and b–f in situ TEM images recorded at 300, 500, 600, 700, and 800 °C, respectively. g SAED pattern corresponding to image a indexed to the orthorhombic structure and h SAED pattern recorded at 500 °C and indexed to the monoclinic WO3 structure
The diffraction pattern shown in Fig. 7h was recorded at 500 °C and was indexed to the monoclinic P21/n (14) WO3 structure. The same structure was identified in all heat-treated samples during the in situ heating experiments in the TEM. The results are in very good agreement with XRD.
One HRTEM image recorded from the extremity of one TNR and its corresponding SAED pattern indexed to the orthorhombic single crystalline structure with the [110] zone axis are shown in Fig. 8a, b. Measurements on the HRTEM images showed that the single crystal TNR growth direction was < 111 >. The measured lattice spacing was 0.35 nm and corresponded to the {111} planes of tungstite. Therefore, the oxolation along equivalent x and y directions ({111} lattice planes) during the crystallization and growth steps leads to the formation of a tungstite crystalline layered phase. Hence, for this layered structure, the z-axis is not in the predominant growth direction. A schematic of one unit cell of tungstite consisting of four distorted octahedra, in accordance with SAED and growth direction, is displayed in Fig. 8c.

HRTEM image of a the tip of a single crystal TNR (as-synthesized). The lattice spacings of 0.35 nm corresponding to the {111} crystal planes (0.35 nm) of the orthorhombic phase and b corresponding SAED pattern showing a zone axis in the [110] direction and reflections pertaining to the orthorhombic structure and c schematic of an orthorhombic tungstite unit cell with growth direction < 111 > . (d–f) HRTEM images recorded in situ at different temperatures (d:300 °C, e:600 °C, and f, g:800 °C) which revealed the presence of oxygen vacancies. Evidence of sintering was not observed. g Higher magnification image recorded from the edge of a monoclinic crystal, with the {110} planes (0.52 nm) labeled and h corresponding SAED pattern indexed to monoclinic WO3 structure with zone axis [001]. i A schematic of a monoclinic WO3 unit cell
The TEM in situ heating experiments resulted in materials that appeared to have a more ordered crystallographic structure on the HRTEM images, as illustrated in Fig. 8d-g. Figure 8h shows a SAED indexed to WO3 with the monoclinic structure recorded at 500 °C (same for all T ≥ 300 °C). A schematic of one unit cell of WO3 consisting of eight distorted octahedra, in accordance with SAED and growth directions, is displayed in Fig. 8i. Therefore, WO3 NRs were 2D monoclinic structures laid in < 200 > directions. The d-spacings were found to be 0.52, 0.52, 0.37, and 0.36 nm corresponding to the (−101), (101), (020), and (200) lattice planes, respectively. The DP shown in (h) was indexed to monoclinic WO3 oriented along the [001] zone axis. Oxygen vacancies were observed in the crystal lattice (see Fig. 8g for example) unlike for heat treatments performed in the ambient atmosphere.
HRTEM images recorded from ex situ heat-treated TNRs in air (i.e., using the tube furnace) are shown in Fig. 9. It resulted in perfect and oxygen vacancy-free monoclinic crystals. O atoms present in air can diffuse to the structure during heating and result in higher levels of crystallinity. A typical SAED pattern, indexed to monoclinic WO3 with zone axis [001], is shown in (f). The weak reflections, e.g., the labeled (110), are forbidden reflections.

HRTEM images recorded from TNR annealed at different temperatures a W300, b W400, c W500, d W700 and e W800, and f typical SAED pattern, indexed to monoclinic WO3 with zone axis [001]
EEL spectra were recorded from samples heated ex situ (samples heated in air) and in situ (samples heated in vacuum). EEL spectra recorded in situ (at RT, 400, 600, and 800 °C) are presented in Fig. 10. The data were smoothed with a 1 eV window. Because they were recorded from one NR at one time, multiple scattering effects can be ignored since the NRs were less than 20 nm thick having little consequence on the energy-loss near-edge structure (ELNES). The O K-edge may be influenced by several factors including the ejection of electrons into the continuum producing a saw-tooth shape, as well as solid-state effects and transitions to unoccupied bound states creating fine structures in the edge [42, 43]. The ELNES of the O K-edge in transition metal oxides is due to the crystal-field splitting of the metallic d orbital into eg and t2g components, resulting in the EELS double-peak signature [44, 45].

EEL spectra recorded in situ showing the O K-edge at different temperatures (RT, 400, 600, and 800 °C) and also recorded from samples WRT1 and WRT2
The peaks positions with respect to the O K-edge are listed in Table SI. The energy for the first peak at high temperatures (400–800 °C) was about 2.0–2.1 eV (with respect to the position of the O K-edge; i.e., 532 eV) and 1.8 eV for sample WRT2. An increase in temperature, therefore, leads to an increase in the level of the energy of the t2g orbital. The energy for the second peak in TNRs (9.01 eV) was higher than that for samples annealed at higher temperatures (400, 600, and 800 °C) which varied from 7.9 to 8.0 eV. The separation between the first and the second peak is higher for TNRs (7.7 eV) than for monoclinic NRs (5.9-6.2 eV). In the orthorhombic structure, there is a strong double bond and a weak water molecule bond which are connected to tungsten. These two bonds contribute mostly to the t2g component. The higher intensity of the second peak for TNRs is due to these two bonds. The O–K ELNES for both phases had a similar shape, but differ in position and intensity [45, 46]. The O K-edge can, therefore, be used for the phase identification of these two materials.
In situ heating experiments in the TEM showed that the position of the first peak shifted from 1.30 eV (TNRs at RT) to 2.0–2.1 eV (WO3 at 400–800 °C), respectively. The EEL spectra for ex situ samples were also recorded (see Figures S1 and S2 and Table SII, ESI). Ex situ EELS (see Table SII, ESI) showed that it also decreased from 2.0 to 2.1 to 1.80 eV when cooling to RT, while the first peak remained unchanged (1.3 eV) for both phases. The peak separation for the monoclinic structure did not change significantly (in situ and ex situ).
UV/Vis absorption spectra recorded from annealed TNRs (WO3 NRs) are shown in Fig. 11. For samples annealed at temperatures 600 °C and below (W300, W400, W500, and W600), absorption was significantly more intense than for samples annealed at temperatures 700 °C and above. Two peaks around 245 nm (5.0 eV) and 365 nm (3.4 eV) appeared for TNRs [34]. Two peaks at 325 nm (3.8 eV) and 245 nm (5.0 eV) and a trailing infrared (IR) absorption were observed for samples W300, W400, and W500. The apparent absorption into the IR range is attributed to the scattering of the WO3 NRs and is reasonable considering the near-micrometer scale (in the xy plane) of the NRs. These peaks can reasonably be assigned to the e *g (σ*-bonds) and t *2g (π*-bonds) orbital components (peak separation distance is 10,050 cm−1) [40]. This peak separation was smaller for TNRs (13,500 cm−1) [34]. Therefore, the phase transformation is responsible for changes in the soft UV region (365 nm to 325 nm). Ignoring the electron correlation effects and considering that the transition from a1g, t2g and eg to t *2g is forbidden, the two observed peaks are the results of the electron excitation from t1u occupied orbitals to two unoccupied anti-bonding components e *g and t *2g corresponding to 245 nm and 325 nm photons. The decrease in absorption when the heat treatment temperature was increased from 300 to 500 °C is attributed to the more rapid settling of the NRs because of sintering. With the exception of the more severe sintering, there were no changes observed when heating was increased to 700 °C and higher.

UV/Vis absorption spectra from TNRs to WO3 NRs sintered at various temperatures in ambient air
The current versus voltage (I–V) characteristics for as-synthesized and ex situ heat-treated TNRs were recorded and are shown in Fig. 12. The samples that were heat treated to higher temperatures displayed a lower conductivity, a consequence of the reduced concentration of oxygen vacancies. The inset in Fig. 12b shows the two plots at the same scale. Therefore, it may be possible to use these materials in desirable applications (e.g., resistive switching [47] ). According to the electrode separation of 20 µm for interdigitated Pt/PtO electrodes, the breakdown electric field value for TNRs was measured to ~3x105 V.m−1, which increased to ~4x105 V.m−1 for sample W300. In all cases, the Schottky barrier behavior was observed. WO3 nanoribbons grafted with Pt nanoparticles (i.e., co-catalyst) are suggested [48] and will be the subject of a future publication.

I–V plot recorded for TNRs (a), W300 and W800 (b), and the inset shows two I-V curves at the same y-axis (current) scale. Inset in a is a photograph of the interdigitated Pt/PtO electrode device used for the measurements
Conclusion
In situ heating experiments in the TEM and temperature-dependent Raman spectroscopy demonstrated the phase transformation of orthorhombic tungstite NRs to monoclinic tungsten oxide NRs, initiated between 200 °C and 300 °C. EELS and XPS showed the electronic structure and bonding properties for tungstite and tungsten oxide NRs. EEL spectra can be used for phase identification between the two materials. Annealing in ambient atmosphere got rid off oxygen vacancies resulting in better stoichiometry but reduced conductivity. In order to avoid sintering and a loss of the advantageous nanoribbon morphology, it is preferable to anneal at temperatures not to exceed 500 °C.
References
Chatten R, Chadwick AV, Rougier A, Lindan PhJD (2005) The oxygen vacancy in crystal phases of WO3. J Phys Chem B 109(8):3146–3156
Soultanidis N, Zhou W, Kiely CJ, Wong MS (2012) Solvothermal synthesis of ultrasmall tungsten oxide nanoparticles. Langmuir 28:17771–17777
Santato C, Odziemkowski M, Ulmann M, Augustynski J (2001) Crystallographically oriented mesoporous WO3 films: synthesis, characterization, and applications. J Am Chem Soc 123:10639–10649
Guo Ch, Yin Sh, Huang Y, Dong Q, Sato T (2011) Synthesis of W18O49 nanorod via ammonium tungsten oxide and its interesting optical properties. Langmuir 27:12172–12178
Zheng H, Tachibana Y, Kalantar-zadeh K (2010) Dye-sensitized solar cells based on WO3. Langmuir 26(24):19148–19152
Hara K, Zhao ZhG, Cui Y, Miyauchi M, Miyashita M, Mori Sh (2011) Nanocrystalline electrodes based on nanoporous-walled WO3 nanotubes for organic-dye-sensitized solar cells. Langmuir 27:12730–12736
Costa C, Pinheiro C, Henriques I, Laia CAT (2012) Inkjet printing of sol-gel synthesized hydrated tungsten oxide nanoparticles for flexible electrochromic devices. ACS Appl Mater Interfaces 4:1330–1340
Wood V, Panzer MJ, Halpert JE, Caruge J-M, Bawendi MG, Bulovic V (2009) Selection of metal oxide charge transport layers for colloidal quantum dot LEDs. ACS Nano 3(11):3581–3586
Srinivasan A, Miyauchi M (2012) Chemically stable WO3 based thin-film for visible-light induced oxidation and superhydrophilicity. J Phys Chem C 116:15421–15426
Yang J, Wang D, Han H, Li C (2013) Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc Chem Res 46:1900–1909
Ahmed S, Hassan IAI, Roy H, Marken F (2013) Photoelectrochemical transients for chlorine/hypochlorite formation at “roll-on” nano-WO3 film electrodes. J Phys Chem C 117:7005–7012
Liu X, Wang F, Wang Q (2012) Nanostructure-based WO3photoanodes for photoelectrochemical watersplitting. Phys Chem Chem Phys 14:7894–7911
Chen X, Ye J, Ouyang Sh, Kako T, Li Zh, Zou Zh (2011) Enhanced incident photon-to-electron conversion efficiency of tungsten trioxide photoanodes based on 3D-photonic crystal design. ACS Nano 5(6):4310–4318
Walter MG, Warren EL, McKone JR, Boettcher ShW, Mi Q, Santori EA, Lewis NS (2010) Solar water splitting cells. Chem Rev 110:6446–6473
Maeda K, Higashi M, Lu D, Abe R, Domen K (2010) Efficient nonsacrificial water splitting through two-step photoexcitation by visible light using a modified oxynitride as a hydrogen evolution photocatalyst. J Am Chem Soc 132:5858–5868
Higashi M, Abe R, Ishikawa A, Takata T, Ohtani B, Domen K (2008) Z-scheme overall water splitting on modified-TaON photocatalysts under visible light (λ<500 nm). Chem Lett 37(2):138–139
Liu Y, Shrestha S, Mustain WE (2012) Synthesis of nanosize tungsten oxide and its evaluation as an electrocatalyst support for oxygen reduction in acid media. ACS Catal 2:456–463
Waller MR, Townsend TK, Zhao J, Sabio EM, Chamousis RL, Browning ND, Osterloh FE (2012) Single-crystal tungsten oxide nanosheets: photochemical water oxidation in the quantum confinement regime. Chem Mater 24:698–704
Coridan RH, Shaner M, Wiggenhorn C, Brunschwig BS, Lewis NS (2013) Electrical and photoelectrochemical properties of WO3/Si tandem photoelectrodes. J Phys Chem C 117:6949–6957
Su J, Guo L, Bao N, Grimes CA (2011) Nanostructured WO3/BiVO4 heterojunction films for efficient photoelectrochemical water splitting. Nano Lett 11:1928–1933
Wang A, Zhang T (2013) Functionalized silica nanoparticles: a platform for fluorescence imaging at the cell and small animal levels. Acc Chem Res 2013(46):1377–1386
Cui X, Shi J, Chen H, Zhang L, Guo L, Gao J, Li J (2008) Platinum/mesoporous WO3 as a carbon-free electrocatalyst with enhanced electrochemical activity for methanol oxidation. J Phys Chem B 112:12024–12031
Li Zh, Smid B, Kim YK, Matolin V, Kay BD, Rousseau R, Dohnalek Z (2012) Alcohol dehydration on monooxo W=O and dioxo O=W=O species. J Phys Chem Lett 3:2168–2172
Li D, Wu G, Gao G, Shen J, Huang F (2011) Ultrafast coloring-bleaching performance of nanoporous WO3-SiO2 gasochromic films doped with Pd catalyst. ACS Appl Mater Interfaces 3:4573–4579
Filho AGS, Filho JM, Freire VN, Ayala AP, Sasaki JM, Freire PTC, Melo FEA, Juliao JF, Gomes UU (2001) Phase transition in WO3 microcrystals obtained by sintering process. J Raman Spectrosc 32:695–699
Salje E, Viswanathan K (1975) Physical properties and phase transitions in WO3. Acta Cryst A31:356–359
Salje E (1977) The orthorhombic phase of WO3. Acta Cryst B33:574–577
Hirose T (1980) Structural phase transitions and semiconductor-metal transition in WO3. J.Phys Soc Jpn 49(2):562–568
Ramana CV, Utsunomiya S, Ewing RC, Julien CM, Becker U (2006) Structural stability and phase transitions in WO3 thin films. J Phys Chem B 110:10430–10435
Vemuri RS, Kamala Bharathi K, Gullapalli SK, Ramana CV (2010) Effect of structure and size on the electrical properties of nanocrystalline WO3 films. ACS Appl. Mater. Interfaces 2(9):2623–2628
In J, Yoo Y, Kim JG, Seo K, Kim H, Ihee H, Oh SH, Kim B (2010) In situ TEM observation of heterogeneous phase transition of a constrained single-crystalline Ag2Te nanowire. Nano Lett 10:4501–4504
Delalande M, Guinel M, Allard LF, Delattre A, Bris R, Samson Y, Guillemaud P, Reiss P (2012) L10 Ordering of ultrasmall FePt nanoparticles revealed by TEM in situ annealing. J Phys Chem C 116:6866–6872
Supothina S, Seeharaj P, Yoriya S, Sriyudthsak M (2007) Synthesis of tungsten oxide nanoparticles by acid precipitation method. Ceram Int 33:931–936
Ahmadi M, Guinel M (2014) Synthesis and characterization of tungstite (WO3·H2O) nanoleaves and nanoribbons. Acta Mater 69:203–209
Merchan WM, Farahani MF, Rosenberg ZM (2014) Electron beam induced formation of tungsten sub-oxide nanorods from flame-formed fragments. Micron 57:23–30
Egerton RF, Li P, Malac M (2004) Radiation damage in the TEM and SEM. Micron 35(6):399–409
Wolcott A, Kuykendall TR, Chen W, Chen Sh, Zhang JZ (2006) Synthesis and characterization of ultrathin WO3 nanodisks utilizing long-chain. J Phys Chem B 110:25288–25296
Szymanski JT, Roberts AC (1984) The Crystal Structure of Tungstite, WO3·H2O. Canadian Minerologist 22:681–688
Loopstra BO, Boldrini P (1966) Neutron diffraction investigation of WO3. Acta Cryst 21:158–162
A.F. Holleman, E. Wiberg, N. Wiberg, “Inorganic chemistry”, Academic Press; Berlin; New York: Walter De Gruyter, San Diego, 2001, p.1389.
Baek Y, Yong K (2007) Controlled growth and characterization of tungsten oxide nanowires using thermal evaporation of WO3 powder. J Phys Chem C 111(3):1213–1218
Sefat AS, Amow G, Wu MY, Botton GA, Greedan JE, Solid J (2005) High-resolution EELS study of the vacancy-doped metal/insulator system, Nd1-xTiO3, x=0 to 0.33. J Solid State Chem 178(4):1008–1016
Ahn CC, Rez P (1985) Inner shell edge profiles in electron energy loss spectroscopy. Ultramicroscopy 17:105–115
Jollet F, Petit T, Gota S, Thromat N, Soyer Gautier M, Pasturel A (1997) The electronic structure of uranium dioxide: an oxygen K-edge x-ray absorption study. J Phys 9:9393–9401
Harvey A, Guo B, Kennedy I, Risbud S, Leppert V (2006) A systematic study of the oxygen K edge in the cubic and less common monoclinic phases of the rare earth oxides (Ho, Er, Tm, Yb) by electon energy loss spectroscopy. J Phys 18:2181–2189
McComb DW (1996) Bonding and electronic structure in zirconia pseudopolymorphs investigated by electron energy-loss spectroscopy. Phys Rev B 54:7094–7120
Kozicki MN, Gopalan C, Balakrishnan M, Mitkova M (2006) A low-power nonvolatile switching element based on copper-tungsten oxide solid electrolyte. IEEE Trans Nanotechnol 5(5):535–544
Abe R, Takami H, Murakami N, Ohtani B (2008) Pristine simple oxides as visible light driven photocatalysts: highly efficient decomposition of organic compounds over platinum-loaded tungsten oxide. J Am Chem Soc 130:7780–7781
Acknowledgements
The partial support from NSEC Center for Hierarchical Manufacturing at the University of Massassuchets (National Science Foundation award 1025020). NSF for its support (award 0701525) to the Nanoscopy Facility, an electron microscopy facility at UPR.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ahmadi, M., Sahoo, S., Younesi, R. et al. WO3 nano-ribbons: their phase transformation from tungstite (WO3·H2O) to tungsten oxide (WO3). J Mater Sci 49, 5899–5909 (2014). https://doi.org/10.1007/s10853-014-8304-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8304-2