
Abstract
A series of organ-soluble copolyimides (co-PIs) were synthesized from 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA), 2,2′-bis(trifluoromethyl)-4, 4′-diaminobiphenyl (TFMB), and 2-(4-aminophenyl)-5-aminobenzimidazole (BIA) via a one-step polymerization in N-methyl-2-pyrrolidone (NMP). The polyimide solutions were used to fabricate as-spun polyimide fiber by a wet-spinning process. SEM images of the round cross-section of the fibers indicated that a homogeneous and dense fibrous structure was produced in the coagulation bath of H2O/NMP = 90/10 (v/v) and many microfibrils appeared on the surface. The drawn fibers exhibit excellent mechanical properties, and the strength and modulus of BTDA/TFMB/BIA co-PI fibers (TFMB/BIA = 50/50) reached 2.25 and 102 GPa with a draw ratio of 3.0. The 5 % weight loss temperature of the co-PI fibers in thermogravimetric analysis spectra achieved 548–563 °C in an air atmosphere. The glass transition temperatures were found to be between 340 and 366 °C according to the DMA results. Annealed BTDA/TFMB/BIA co-PI fibers displayed distinct wide-angle X-ray patterns, and crystallinity and crystal orientation with various draw ratios were observed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As an important member of aromatic heterocyclic polymers, polyimide (PI) fibers exhibit many valuable properties such as outstanding thermal stability, chemical resistance, and good resistance to irradiation [1–4], resulting in their extensive applications in engineering plastics and composite materials. However, high-strength polyimide fibers have not been produced in large scale. Two main techniques have been well-developed to prepare the fibers [4–6]. One is two-step process: soluble poly(amic acid) (PAA) in a polar solvent, the precursor of polyimide, is firstly spun into a coagulation bath to prepare PAA fibers, and then the PAA fibers are converted into infusible and insoluble PI fibers via thermal or chemical imidization. Alternatively, the precursor fibers can be prepared by dry-spinning method, in which the dope is spun into a long heating tube [7]. This spinning technology has been considered to be promising for realizing industrial level production of PI fibers. However, the imidization process of PAA into polyimide is a complicated reaction, and has a negative effect on the microstructure and properties of the fiber, due to evaporation of residual solvent and a trace of water produced in the imidization reaction. The other process is the one-step spinning technology, in which soluble polyimide in a solvent is spun into PI fibers directly, no the precursor fibers appear and the imidization process is ignored thus. Kaneda et al. [5, 8] reported that polyimide solutions were prepared in the one-step polycondensation reaction of a dianhydride with various aromatic diamines in p-chlorophenol, and the dopes were directly spun into polyimide fibers. The spinning and drawing procedures were carried out on a continuous line. Cheng and co-workers [9, 10] synthesized a series of soluble polyimides in m-cresol and the fibers were prepared by one-step spinning technology, and the strength and modulus of the fibers reached 3.1 and 130 GPa, respectively. However, the limitation of monomer selection for organ-soluble polyimides and the toxicity of the employed phenol solvents severely hinder the larger-scale production of the fibers. To overcome these problems, much effort has been focused on the synthesis of soluble polyimide in friendly organic solvents such as polyphosphoric acid (PPA) [11], benzoic acid (BA) [12], and N-methyl-2-pyrrolidone (NMP) [13]. Several successful approaches to soluble polyimides have been developed, including insertion of flexible linkage or bulky substituents on the main chains and utilization of noncoplanar monomers [14, 15].
Copolymerization with two different diamine or dianhydride monomers is also known as an effective way to improve the solubility of polyimides [6]. The heterocyclic diamine 2-(4-aminophenyl)-5-aminobenzimidazole (BIA) is a versatile monomer with an asymmetry structure feature, which has strong inter- or intra-molecular hydrogen-bonding interaction that may strengthen the mechanical properties of resulting materials. It has been successfully introduced into rigid-rod, para-substituted, wholly aromatic polyamide fibers (armid-fiber, Armos) in order to improve the solubility and drawability. Liu et al. [17] prepared a series of co-PI fibers containing BIA units by two-step process and the strength and modulus of the fibers reached 1.53 and 220 GPa, respectively. Introduction of this unit into the polymer chains will create intermolecular interaction, which has a great effect on mechanical properties of the fibers. The diamine, 2,2′-bis-(trifluoromethyl)-4,4′-diaminobiphenyl (TFMB), possesses a unique structure, a rigid nonplanar structure with two bulky trifluoromethyl groups [15, 16]. Cheng et al. [2, 9, 10, 18] synthesized a series of soluble polyimides containing this monomer using m-cresol as the solvent and fabricated high performance polyimide fibers. They reported that the introduction of bulky CF3 groups into the macromolecular chain resulted in great benefits for improving solubility of polyimides. However, the unpleasant smell and toxicity of the m-cresol limited the mass production of those fibers.
Solubility and spinnability of polyimides are the important issues for preparing high polyimide fibers. Here, three monomers, 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA), TFMB, and BIA, were used to synthesize copolyimides, and the resulting copolyimides exhibited an excellent solubility in NMP due to the introduction of BIA and TFMB in the macromolecular chains. Then, one-step spinning technology was employed to prepare the fibers, and the drawn fibers showed good mechanical properties.
Experimental
Materials
BTDA was obtained from Beijing Multi Technology co., Ltd., and dried in vacuum at 120 °C for 24 h prior to use. TFMB and BIA were purchased from Changzhou Sunlight Pharmaceutical co., Ltd. NMP was stirred in the presence of P2O5 overnight and then distilled under reduced pressure. Other commercially available reagent grade chemicals were used without further purification.
Characterization
Inherent viscosities (η inh) were obtained on 0.5 g/dL concentration with an Ubbelohde viscometer in NMP at 30 °C. ATR-FTIR spectra of the polyimide fibers were obtained with a Nicolet 8700 spectroscope in the range of 4000–400 cm−1. 1H NMR spectra was recorded on Bruker Avance 400 nuclear magnetic resonance spectrometers, using DMSO-d6 as solvents and tetramethylsilane as an internal reference. Differential scanning calorimetry (DSC) was performed on a Netzsch DSC 204F1 thermal analyzer system at a heating rate of 3 °C/min under nitrogen atmosphere. Thermogravimetric analysis (TGA) was conducted on a Discovery TGA Q5000IR. The measurement of linear coefficients of thermal expansion (CTE) and dynamic mechanical behavior of the co-PI fibers were carried out on a thermomechanical analyzer (TA, Q800). The mechanical properties were measured using Donghua XQ-1 instrument. Morphologies of the cross-section of the fibers were observed on a scanning electron microscope (SEM) (HITACHI SU8010) at an accelerating voltage of 3.0 kV. X-ray diffraction (XRD) measurements were carried out using a Rigaku D-max-2550 diffractometer with CuKα radiation. The crystallinity was calculated using powder diffraction patterns obtained from chopped fibers between 0° and 360°. Crystal orientation factor was obtained through an azimuthal integration of a preferred crystalline plane with the clearest diffraction along the c axis using a normal beam direction with respect to the fiber sample.
Synthesis of BTDA/TFMB/BIA polyimides
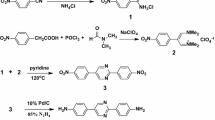
Equimolar amounts of diamine and dianhydride monomers were used in all cases and copolyimide solutions in NMP were produced. Polymerization was carried out with various ratios of TFMB to BIA: 85/15 (co-PI-1), 75/25 (co-PI-2), 50/50 (co-PI-3), and 40/60 (co-PI-4). The reaction was shown in Schemes 1, 2, and a representative polymerization was as follows: A 250 mL three-necked flask equipped with a nitrogen inlet and a mechanical stirrer was charged with distilled NMP and appropriate amount of TFMB and BIA. Once the diamines were dissolved, the equimolar dianhydride was added. The solution was stirred at room temperature for 3 h, and isoquinolin (~0.2 g) was added and further stirred for 3 h at 120 °C. Then the mixture kept at 195 °C for 10 h, and water from the imidization reaction was continuously was removed with a stream of nitrogen. Polyimide solution with a concentration of 10 wt% was thus obtained.

Synthetic route of copolyimides (m/n = 85/15, 75/25, 50/50, and 40/60)

Flow process diagram of polyimide fiber by wet spinning. a Co-polyimide solution. b A spinning spinneret with 50 holes. c Coagulation bath with a mixture solution of water and NMP (90/10, v/v). d, e Washing bath, water. f Taking up roller
Preparation of copolyimide fibers
The copolyimide solutions were filtered and degassed at 60 °C prior to spinning. The co-PI fibers were spun by wet-spinning. The co-PI dopes were extruded through a spinneret (50 holes with a diameter of 80 μm) into a coagulation bath. The solidified filament entered into the second and third washing baths with 60 °C water, and then clustered at a taking up roller. The as-spun co-PI fibers were drawn through a heating tube with three temperature parts (450, 480, and 500 °C) under the nitrogen atmosphere. The drawn ratios are controlled by the speeds of two taking up rollers.
Results and discussion
Polymer synthesis
Co-PIs were synthesized by reacting the stoichiometric amounts of a diamine mixture with BTDA via a one-step synthetic method, as shown in Scheme 1 [16]. In other words, no precursor (polyamic acid) was produced in the synthetic process, and co-polyimides were directly prepared in the solution instead. The inherent viscosities of the co-PIs range from 0.82 to 2.80 dL/g. In detail, the values of the co-PIs with the ratio of TFMB to BIA of 85/15, 75/25, 50/50, and 40/60 are 0.82, 1.03, 2.83, and 2.80 dL/g, respectively. Apparently, the increase of TFMB contents results in the decrease of viscosities of polymer solutions, attributing to the lower activity of TFMB owing to the electron-withdrawing effect and the relatively larger steric hindrance of the two bulky trifluoromethyl groups.
As expected, the polyimides from the mixed diamine monomers exhibit sufficient solubility due to the reduced regularity of the repeating unit in spite of the presence of electron-withdrawing CF3 and rigid rodlike structure. The solubility of the co-PIs in various organic solvents is summarized in Table 1. Apparently, the copolymers show an excellent solubility in aprotic polar solvents such as NMP, DMAc, DMF, and DMSO and they are also soluble in less polar solvents such as CHCl3, while they are insoluble in carbon tetrachloride (CCl4) and toluene. The solubility of the co-PI fibers is enhanced due to the introduction of bulky pendant trifluoromethylpheneyl groups which can inhibit close packing and random copolymerization reducing the chain regularity. Meanwhile, we can observe that the solubility of co-PI fibers reduces with the decrease of TFMB content. However, the heat-treated co-PIs are insoluble in any solvent. The poor solubility of the thermally treated polymer is possibly due to the presence of partial intermolecular crossing [19]. The changes of solubility before and after heat treatment can insure the safe use of co-PI fibers.
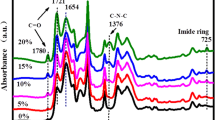
Structures of the co-PI fibers were confirmed by ATR-FTIR. Figure 1 shows the representative ATR-FTIR spectra for the untreated co-PI-3 fiber and that thermally treated at 350 °C. Apparently, the presence of characteristic absorption for carbonyl group of the imide ring at 1780 and 1723 cm−1 and characteristic band for C–N vibration at 1374 and 723 cm−1 confirms the formation of imide rings. Furthermore, the two spectra show the characteristic absorption bands for C–F at about 1150 and 1667 cm−1 corresponding to the N–H stretching in the benzimidazole. No absorption due to the amide bond appears in the as-spun fiber FTIR spectra, and it is ascertained that both polycondensation and imidization are completed in this one-step process.

ATR-FTIR spectra of co-PI-3 fiber
1H NMR spectra of co-PI-3 were provided to further confirm the structure of this polymer. As shown in the inset of Fig. 2, Hi, Hj, and Hk close to the imide ring appear at the higher field region of the spectrum because of the resonance. The proton Hm corresponding to benzimidazole-ring appears at the farthest downfield region. As expected, the 1H NMR spectra do not show any amide and acid protons, indicating full imidization of the polymer. Moreover, no imidization transition in TGA and DSC results implies that the cyclization reaction in solution fully completes.

1H NMR spectra of co-PI-3 fiber
Morphology of co-PI fibers
Figure 3 shows the cross-section and surface morphology of as-spun co-PI fibers produced from various compositions of H2O/NMP mixtures. As for as-spun fiber in Fig. 3(a1), the cross-section of the fiber coagulated in pure water is ellipse shape without macrovoids. Figure 3(a2) shows that some gel particles and macrovoids appear on the surface of the fiber. During wet spinning, the coagulation of fibers is a dual diffusion process, namely, solvent NMP diffusing into the bath and the non-solvent water diffusing into the fibers. The diffusion first takes place around the surface of the fibers, and the surface “skin” is formed, which baffles the diffusion of water into the core of the fibers, so the surface is compact and the core is loose [20–22]. When pure water is used as the coagulant, the concentration grads is so much that the inner force and out force cannot reach equilibrium, leading to the irregular and asymmetric cross-section. As increasing the NMP content in the coagulation bath, the cross-sectional shape of the fiber deviates more from an ellipse shape, showing a circular form in a composition of water/NMP (90/10). Alternatively, the cross-section of the fiber is round and voids free, and the “skin–core” structure is not obvious, as shown in Fig. 3(c1). The surface of the fiber shows a large number of microfibrils along the longitudinal axis. The results mean that this coagulation of water/NMP (90/10) is very beneficial to forming homogeneous and dense fibrous structure.

Cross-section and surface morphology of co-PI fibers produced from various compositions of coagulation bath of H2O (a1, a2), H2O/NMP = 95/5 (v/v) (b1, b2), and H2O/NMP = 90/10 (c1, c2)
Crystallinity and crystal orientation of the fibers
In order to study the crystallinity and crystal orientation in the fibers, X-ray diffraction of the fibers was carried out. Figure 4a shows X-ray patterns of co-PI-3 fibers with various draw ratio from 1.5 to 3.5 times. It is evident that the curve of the as-spun fiber is broad and no obvious peak appears, indicating an amorphous state. In general, unstretched polyimides exhibit amorphous state, such as films or engineering plastics. Here, the polymer backbones with multiple trifluoromethyl groups result in the reduction of the chain–chain interaction and the decrease of the chain packing efficiency and thus hinder the polymer crystallization [13]. However, two strong reflections at 12.3° and 16.1° are observed in the X-ray diffraction of the drawn fibers. The intensity of reflection peaks enhances upon increasing draw ratios, demonstrating that the tensile stress induces the crystallinity and crystal orientation of the fibers.

X-ray diffraction of co-PI-3 fibers at various draw ratios (a), and dependences of crystallinity orientation factor on draw ratios (b)
The crystallinity determination of the drawn-fiber was conducted through a powder pattern method of the chopped fibers. Figure 4b illustrates the relationship between the crystallinity and draw ratio. The crystallinity increases from 20 % at λ = 1.5 to 38 % at λ = 3.0. While, this increasing trend is restricted at a high draw ratio (>3.0), which may be attributed to the fact that the excessive drawing-treatment destroys the integrity of the crystalline region of the material. As reported by Eashoo et al. [10], crystal orientation factor can be obtained through an azimuthal integration of a preferred crystalline plane along the c axis using a normal beam direction with respect to the fiber sample. The orientation factor was calculated from Hermans orientation function:
where f c is the orientation factor along the fiber direction, and ϕ c represents the angel between the center diffraction of the preferred crystalline plane and the c axis of the crystal unit cell. The numerical values of the mean-square cosines in this equation can be determined from the fully corrected intensity distribution reflected from this preferred crystalline plane, I c (ϕ c, α), averaged over the entire surface of the orientation sphere:
According to the above expression, the change of preferred orientation factor of the co-PI fibers with various draw ratios can be obtained, appeared in Fig. 4b. The orientation factor of the heat-draw at λ = 1.5 is about 0.76, and it increases to 0.84 at a draw ratio of 3.0. This result indicates that the drawing stress induces the orientation of the crystals, which benefits to the improvement of mechanical properties of these fibers.
Mechanical properties of the co-PI fibers
The representative plots of stress versus strain of the fibers are shown in Fig. 5, and the mechanical properties of the drawn co-PI-3 fibers are listed in Table 2. For as-spun fibers, the tensile to break is around 0.36 GPa with an elongation of up to 27 %. The drawing process remarkably enhances the tensile strength of the fibers. The value reaches 1.58 GPa at λ = 2.0 and 2.25 GPa at λ = 3.0. Correspondingly, the strain to break decreases from 12 % at λ = 2.0 to 5.4 % at λ = 3.0.

Typical stress–strain diagrams for co-PI-3 fibers with various draw ratios
The initial modulus shows a drastic increase as the draw ratio increases. As-spun fiber has an initial modulus of 14 GPa, the drawn fiber with λ = 2.0 exhibits an enhanced modulus of 37 GPa and then the value reaches 102 GPa at λ = 3.0. This behavior is different from those fibers spun from liquid–crystalline states, such as Kevlar fibers [23, 24] and PBO fibers [25, 26]. In those cases, the as-spun fibers have relatively high strength and modulus. Since the BTDA/TFMB/BIA fibers are spun from an isotropic solution rather than a lyotropic liquid–crystalline state, the macromolecules are not well oriented and non-crystallinity in the as-spun fibers. As a result, the fibers undergo considerable elongation during drawing process. The advantage of this spinning method is that, during drawing process, the macromolecular chains have much opportunity to rearrange themselves to defect-free positions under the large deformation.
Thermal stability and dynamic mechanical behavior
The thermal properties of co-PI fibers were evaluated by DSC and TGA. DSC curves in Fig. 6 demonstrate the high glass transition temperature (T g) due to the rigid rod-like characteristics of the fibers. The T gs of the co-polyimides are 340, 355, and 358 °C for co-PI-1, co-PI-2, and co-PI-3, respectively. While, co-PI-4 fiber shows no distinctly T g at determined temperature region. The increment of T g with more BIA content is attributed to the increase of chain rigidity and the hydrogen bonding between the N–H groups of benzimidazole ring and the C=O groups of imide ring [27, 28].

DSC curves of the co-PI fibers at a heating rate of 5 °C/min
The thermal stabilities of the co-PI fibers were evaluated by TGA measurement in both nitrogen and air atmospheres. As listed in Table 3, the co-PI-3 fiber in nitrogen is thermally stable up to 590 °C at a 5 % weight loss, while the value is 530 °C in air atmosphere. All samples show similar pyrolysis behaviors in N2. The T 5 % values of co-PI-1, co-PI-2, co-PI-3, and co-PI-4 fibers are 591, 560, 539, and 530 °C. The 55–60 % of the original mass retains even at a high temperature of 750 °C, indicating a high carbon yield for the materials. These results indicate that the co-PI fibers exhibit excellent thermal stability and antioxidation property.
The dynamic mechanical behaviors of the heat-treated co-PI-3 fibers are investigated, as shown in Fig. 7a. The heat-treated fiber exhibits two relaxation transitions above room temperature. (1) A sub-glass relaxation at 270 °C, namely, β relaxation with an activation energy of 209 kJ/mol. The magnitude of this transition is about 0.1 in tanδ scale. This sub-glass transition is similar to other β relaxations in aromatic polyimide [9]. (2) In the high temperature region, the glass transition (α relaxation) occurs at ca. 363 °C, which is consistent with the DSC results. In order to investigate the influence of diamine ratios on glass transition temperatures, DMA measurement of the four samples are carried out. As shown in Fig. 7b, the α relaxation of these fibers is clearly observed at the temperature ranges from 341 to 366 °C. In other word, the increasing BIA content results in a high glass transition temperature. Meanwhile, the maximum values of tanδ exhibit the same trend, from 0.27 to 0.38. It should be noted that α relaxation process corresponds to the segmental motion. The intensity variations of α relaxation represent the energy consumption in chain segments motion. The intermolecular interaction is more stronger among the polymer chains with the increase of BIA content in the polymer chain that affects the chain segment motion, resulting in a relative high glass transition temperature.

Dynamic mechanical curves of heat-treated co-PI-4 fibers (a), and dependence of Tanδ of co-PI fibers on BIA content (b)
The thermal shrinkage behavior is of importance for the high performance fibers. Figure 8 shows the changes of thermal shrinkage strain with the temperature for co-PI-3 fibers at various draw ratios. It is clearly to find that thermal shrinkage strain continuously increase with increasing temperature (<300 °C). The linear coefficient of thermal expansion (CTE) of the fiber at an initial stress of 8 MPa can be obtained from the slope during the shrinkage period. For instance, the CTE is −2.4 × 10−4 °C−1 at a draw ratio of 1.5 times and −2.0 × 10−4 °C−1 at a draw ratio of 3.5.

Thermal shrinkage at a stress of 8 MPa for the fibers with various draw ratios
Conclusion
High-performance co-PI fibers based on BTDA, TFMB, and BIA were successfully prepared via the one-step spinning technology. The copolymers showed the perfect solubility in polar solvents with inherent viscosities of 0.82–2.80 dL/g. The optimum co-PI fibers were obtained in a coagulation bath with a water/NMP mixture of 90/10 (v/v). These fibers were further drawn and annealed above 450 °C to obtain outstanding mechanical properties. The tensile strength and Young’s modulus of the fibers were up to 2.25 and 102 GPa when the draw ratio reached to 3.0 times. The glass transition temperatures were found to ranging from 340 to 360 °C by DSC and DMA measurements, which strongly depended on the ratio of the two diamines. Meanwhile, the co-PI fibers displayed excellent thermal stability in accordance to TGA results. As illustrated in XRD, drawing stress induced the crystallinity and orientation of the crystals, and the mechanical properties were thus enhanced.
References
Zhang QH, Dai M, Ding MX, Chen DJ, Gao LX (2004) Euro Polym J 40:2487
Eashoo M, Shen D, Wu Z, Lee CJ, Harris FW, Cheng SZD (1993) Polymer 34:3209
Gao G, Dong L, Liu X, Ye G, Gu Y (2008) Polym Eng Sci 48:912
Park SK, Farris RJ (2001) Polymer 42:10087
Kaneda T, Katsura T, Nakagawa K, Makino H, Horio M (1986) J Appl Polym Sci 32:3133
Kapantaidakis GC, Koops GH, Wessling M (2002) Desalination 144:121
Xu Y, Wang S, Li Z, Xu Q, Zhang Q (2013) J Mater Sci. doi:10.1007/s10853-013-7310-0
Kaneda T, Katsura T, Nakagawa K, Makino H, Horio M (1986) J Appl Polym Sci 32:3151
Cheng SZD, Wu Z, Mark E, Steven HLC, Frank WH (1991) Polymer 32:1803
Eashoo M, Wu Z, Zhang A, Shen D, Tse C, Harris FW, Cheng SZD, Gardner KH, Hsiao BS (1994) Macromol Chem Phys 195:2207
Sakaguchi Y, Kato Y (1993) J Polym Sci, Part A: Polym Chem 31:1029
Kuznetsov AA (2000) High Perform Polym 12:445
Lin CH, Chang SL, Peng LA, Peng SP, Chuang YH (2010) Polymer 51:3899
Qiu Z, Wang J, Zhang Q, Zhang S, Ding M, Gao L (2006) Polymer 47:8444
Chung CL, Hsiao SH (2008) Polymer 49:2476
Chung IS, Park CE, Ree M, Kim SY (2001) Chem Mater 13:2801
Liu X, Gao G, Dong L, Ye G, Gu Y (2009) Polym Adv Technol 20:362
Cheng SZD, Arnold FE, Zhang A, Hsu SLCand Harris FW (1991) Macromolecules 24:5856
Shao Y, Li YF, Zhao X, Wang XL, Ma T, Yang FC (2006) J Polym Sci, Part A: Polym Chem 44:6836
Dorogy WE, Clair AKS (1993) J Appl Polym Sci 49:501
Dong XG, Wang CG, Bai YJ, Cao WW (2007) J Appl Polym Sci 105:1221
Um IC, Kweon H, Lee KG, Ihm DW, Lee JH, Park YH (2004) Int J Biol Macromol 34:89
Tanner D, Fitzgerald JA, Phillips BR (1989) Angew Chem Int Ed 28:649
Hancock TA, Spruiell JE, White JL (1977) J Appl Polym Sci 21:1227
Ernst B, Denn MM, Pierini P, Rochefort WE (1992) J Rheol 36:289
Chae HG, Kumar S (2006) J Appl Polym Sci 100:791
Xia Q, Liu J, Dong J, Yin C, Du Y, Xu Q, Zhang Q (2012) J Appl Polym Sci 129:145
Liu J, Zhang Q, Xia Q, Dong J, Xu Q (2012) Polym Degrad Stab 97:987
Acknowledgements
Financial support of this study was provided by NSFC (51233001, 51173024), 863 plan (2012AA03A211), and 111 Project (111-2-04).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dong, J., Yin, C., Luo, W. et al. Synthesis of organ-soluble copolyimides by one-step polymerization and fabrication of high performance fibers. J Mater Sci 48, 7594–7602 (2013). https://doi.org/10.1007/s10853-013-7576-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7576-2